
Integrated in-depth bioinformatic analysis suggests RELCH/KIAA1468, LINC02341, and AKAP11 as candidate genes for ages at menarche and menopause
Abstract
Background: Polymorphisms of the TNFRSF11A and TNFSF11 genes were reported for their association with age at menarche (AAM) and age at natural menopause (ANM). However, the biological mechanisms underlying this association remain largely unclear. The aim of the study: This study was to determine biological processes backing the observed genetic associations. Materials and methods: Forty-four SNPs were analyzed using in silico approach and ten publicly available online databases and tools. Results: TNFRSF11A and TNFSF11 are highly pleiotropic genes that play a role in many metabolic processes. However, among that variety, lipid metabolism and cell survival and apoptosis seem the most biologically plausible mechanisms, through which these genes contribute to AAM and ANM. The analysis identified several mechanisms underlying the previously determined association of the TNFRSF11A and TNFSF11 genes with AAM and ANM and suggested RELCH/KIAA1468, LINC02341, and AKAP11 as new candidate genes for the traits. Conclusion: The in silico analysis is a powerful approach making it possible to uncover possible metabolic pathways underlying observed genetic associations.
Introduction. Tumor necrosis factor receptor superfamily, member 11a (TNFRSF11A), also known as receptor activator of nuclear factor-κB (NF-κB; RANK), and its ligand (TNFSF11 or RANKL) have been implicated in various cellular processes related to proliferation and death, immunity, and tissue development. The TNFRSF11A/TNFSF11 system is widely acknowledged as one of the key players in some primary postmenopausal disorders, such as osteoporosis [1] and cardiovascular diseases [2]. Also, these genes are expressed in mammary gland cells and were shown to control the development of a lactating mammary gland during pregnancy [3]; that is, they play a role in the reproductive system. Several candidate gene association studies suggested that TNFRSF11A and TNFSF11 were associated with ages at menarche and menopause in different ethnic populations [4-7]. However, biological mechanisms, which underlie these associations, remain largely unclear. The exponential growth of biomolecular data and its mining into databases have provided not only a possibility of more accurate and substantiated choice of genetic markers for a study but also tools for comprehensive analysis to get deeper insights into probable functional assignments of the candidate genetic variants and mechanisms of their contribution to traits [8-10]. I took advantage of the recent advances in bioinformatics and used several online genomic databases to conduct a comprehensive in silico analysis of the TNFRSF11A and TNFSF11 polymorphisms, which were reported as associated with age at menarche and menopause. This bioinformatic analysis aimed to get insights into possible mechanisms of these associations.
Materials and Methods
Selection of polymorphisms
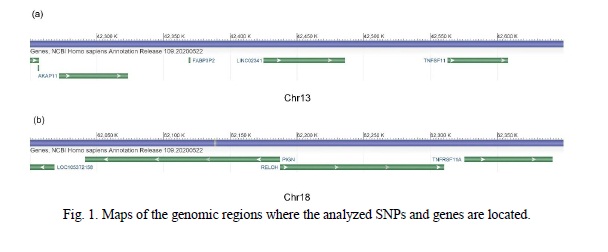
Polymorphisms for the analysis were selected based on the published results of their association with ages at menarche and/or menopause. For this purpose, PubMed was screened using terms “TNFRSF11A”, “TNFSF11”, “RANK”, “RANKL”, “menarche”, and “menopause” in various combinations. The search returned four articles with relevant results. These articles reported in total 44 SNPs (reference polymorphisms hereafter) associated with ages at menarche and/or menopause in three ethnic samples: Caucasians, Chinese, and Mexicans. The list of the selected polymorphisms and the map of the genomic regions, in which they are located, are given in Table 1 and Figure 1.


Bioinformatic analysis
In total ten bioinformatics tools were employed for the analyses.
The effect of non-synonymous SNPs on the protein function was analyzed using SIFT (https://sift.bii.a-star.edu.sg/) [11].
The integrated online tool, HaploReg v4.1 [12] was used to identify polymorphisms in strong linkage disequilibrium (LD) (r2 ≥ 0.8) with the AAM- and/or ANM-associated ones and to analyze them for their functional significance (chromatin states, motifs changes, protein interactions, regulatory potential, and eQTLs). The analysis was conducted separately for Caucasian and Chinese ethnicities using the data of the European and Asian populations from the 1000 Genomes Project Phase.
In addition to HaploReg (v4.1), three other databases were used to analyze regulatory effects of the polymorphisms: RegulomeDB (Version 1.1) (http://regulome.stanford.edu/) [13], rSNPBase (http://rsnp.psych.ac.cn/index.do) [14], and SNP Function Prediction (FuncPred) (https://snpinfo.niehs.nih.gov/snpinfo/snpfunc.html) [15], and GeneCards (https://www.genecards.org/) [16].
The effect of the 44 candidate SNPs for AAM and ANM on gene expression level (cis- and trans-eQTL) was estimated in peripheral blood using the Blood eQTL browser (http://genenetwork.nl/bloodeqtlbrowser/) [17], and in other organs and tissues using the GTExportal data (http://www.gtexportal.org/) as of 07/27/2020. The false discovery rate (FDR) ≤0.05 was applied as the significance level.
The functional significance of the candidate genes for AAM in the various biological pathways was studied using the Gene Ontology Resource tools available at http://geneontology.org [18]. The results of multiple comparisons were adjusted with the FDR<0.05. The gene interaction networks were constructed using GeneMANIA (version 3.5.0) [19] available at http://genemania.org.
Results
Genomic location of the SNPs
First of all, 21 out of the 44 reference SNPs previously annotated to the regions of the TNFRSF11A and TNFSF11 genes could also be mapped to the regions of the other genes (Table 1). Ten reference SNPs were located in the region of the LINC02341 gene, five of them in the introns. Nine variants were located in the 3’-UTR of the AKAP11 gene. Two SNPs, rs2981003 and rs2981004, were located in the 3’-UTR of the RELCH/KIAA1468 gene.
Non-synonymous SNPs
Only one of all analyzed SNPs, rs1805034 in the TNFRSF11A gene, was missense. It results in an Ala/Val replacement in the respective protein. The replacement has SIFT Score = 1 and prediction value “tolerated”.
SNPs in strong LD with the reference polymorphisms
The query against the HaploReg database returned in a total of 348 (224 unique) SNPs linked to the reference ones of the TNFRSF11A gene and 779 (322 unique) SNPs linked to the reference loci of the TNFSF11 gene (Supplementary Table 1). The SNP association and linkage patterns were quite different between European and Asian populations. Specifically, two SNPs of the TNFRSF11A gene, rs3826620 and rs8086340, were associated with AAM and/or ANM in both Caucasians and Chinese [6, 7]. However, the HaploReg analysis returned no SNPs linked to rs3826620 in Europeans vs eight SNPs in Asians. In total, six loci were linked to the three reference SNPs in the European population and 218 were linked to the 19 reference SNPs in the Asian population. Out of these 224 unique SNPs, only three were shared between the European and Asian populations. Quite a few SNPs in the Asian population were located at/near the RELCH/KIAA1468 and PIGN genes (Supplementary Table 1).
Even more striking ethnicity-related differences were observed for the TNFSF11 gene polymorphisms: no shared SNPs in Europeans and Asians. In the Asian population, more than half reference and linked to them polymorphisms were located at/near the AKAP11 gene (Supplementary Table 1).
Regulatory effects
The results of the regulatory effect analysis are shown in Supplementary tables 1 and 2. They suggest that all reference SNPs can produce various regulatory effects, albeit to a different extent. For example, rs8086340 of the TNFRSF11A gene displays histone marks associated with promoters in six tissues and enhancers in 14 tissues, located in the DNase-1 hypersensitive region in 21 tissues, binding region for six proteins, and altered motif for the Foxm1 transcription factor (Supplementary Table 1).
Expression QTLs
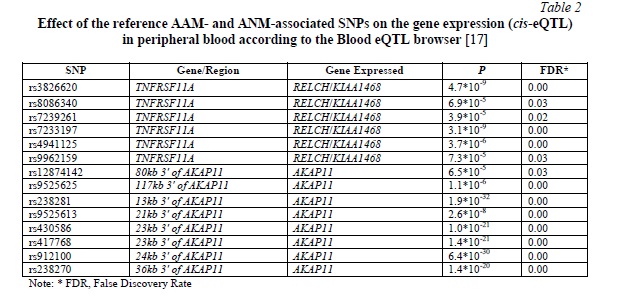
Several reference SNPs appeared to have a significant cis-eQTL effect on the expression of five genes, RELCH/KIAA1468, PIGN, AKAP11, TNFRSF11A, and TNFSF11, in various tissues and organs (Tables 2 and 3).
Pathway analysis
This analysis was conducted for TNFRSF11A and TNFSF11 (because they were originally reported as associated with AAM and/or ANM), LINC02341 (because several reference polymorphisms were mapped to this gene), and RELCH/KIAA1468, PIGN, AKAP11 (because the expression of these genes might be affected by some reference SNPs according to the eQTL analysis).
According to the results of the PANTHER overrepresentation test, the TNFRSF11A and TNFSF11 genes are involved in a broad range of biological processes, including regulation of ERK1 and ERK2 cascade, secretion of prostaglandins, bone remodeling, and mammary gland development (Supplementary Table 3). Apart from these two, AKAP11 was suggested to contribute to the organism's homeostasis (Supplementary Table 3). No data was found for RELCH/KIAA1468, PIGN, and LINC02341.


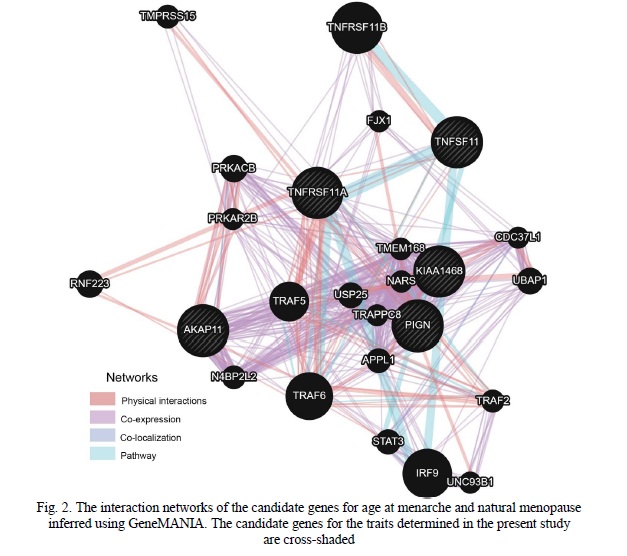
The gene-gene interaction network inferred using GeneMANIA (Figure 2) suggested that the major contribution (64.32%) came from physical interactions between the proteins, followed by co-expression (25.88%), co-localization (5.61%), and common pathways (4.19%).
Discussion. This study provides evidence that, in addition to the TNFRSF11A and TNFSF11 genes previously reported as associated with AAM and/or ANM, four other genes might be associated with these traits.
The LINC02341 gene belongs to the class of long non-coding RNAs. There is not much information about LINC02341 in public databases. Although long non-coding RNAs have not been studied well, there is a growing body of evidence that they are involved in transcriptional regulation [20]. Indeed, according to the GeneHancer database [21], LINC02341 harbors enhancers for six genes, including TNFSF11 and AKAP11, and binding sites for 76 transcription factors. The expression of the gene is relatively low and was documented in several tissues and organs, including lymph nodes, kidneys, placenta, and others [22]. The reference SNP, rs9525625, which is an intronic variant of the gene, was reported as a risk factor of inflammatory bowel disease [23].
The regions of two genes, RELCH/KIAA1468 and AKAP11, also harbored several reference SNPs associated with AAM (Table 1). Besides, quite a few genetic variants in these genes were linked to the reference polymorphisms (Supplementary Table 1). These results suggested that the above genes might also contribute to the above trait.
RELCH (RAB11 binding and LisH domain, coiled-coil and HEAT repeat-containing, alias KIAA1468) encodes a protein playing a key role in intracellular cholesterol distribution [24]. The gene is ubiquitously expressed in human tissues and organs, including endocrine glands, endometrium, and ovaries [22]. The results of the GeneMANIA analysis suggested that this gene was co-expressed with TNFRSF11A, AKAP11, and PIGN (Figure 2).
A product of AKAP11, A-kinase anchoring protein 11, belongs to the protein family whose members, despite the diverse structure, have the same function of binding to the regulatory subunit of protein kinase A and targeting the enzyme to specific locations in the cell. It has similar to RELCH expression patterns [22] but is not co-expressed with TNFRSF11A and PIGN (Figure 2).
The PIGN gene encodes ethanolamine phosphate transferase, a key element of glycosylphosphatidylinositol-anchor biosynthesis. Mutations in the gene were associated with multiple congenital anomalies-hypotonia-seizures syndrome [25]. The gene is co-expressed with TNFRSF11A and RELCH (Figure 2).
RELCH and AKAP11 are pleiotropic genes and were associated with multiple traits, including those related to menarche and menopause (e.g., bone phenotypes, obesity, development, etc.) [26, 27]. There is ample evidence that the above phenotypes have a shared genetic basis with AAM and ANM (see e.g., [6, 7, 28]. Together with the results of the in silico analysis of the present study, it suggests that RELCH/KIAA1468, LINC02341, and AKAP11 may be candidate genes for AAM and/or ANM. This assumption is biologically plausible too.
A possible contribution of PIGN to AAM and/or ANM looks less obvious, largely due to the lack of data about the association of this gene with menarche- and menopause-related phenotypes. On the other hand, according to GeneHancer, this gene harbors binding sites of multiple transcription factors targeting the expression of RELCH and TNFRSF11A. Furthermore, given the involvement of this gene in the basic cellular and developmental processes [29] and tight linkage to the AAM-associated loci (Supplementary Table 1), the above possibility could not be ruled out.
The results of the Gene Ontology and GeneMANIA analyses (Supplementary tables 3, 4, Figure 2) suggested that the contribution of TNFRSF11A and TNFSF11 to menarche and menopause timing is likely multifaceted. The TNFRSF11A/TNFSF11/TNFRSF11B (RANK/RANKL/OPG) signaling pathway has been widely acknowledged as a key player in bone remodeling [1]. Apart from this, the system plays an important role in the progesterone-driven proliferation of the mammary gland epithelium and the risk of breast cancer [30]. One of the possible ways through which TNFRSF11A can affect AAM and ANM is an interaction with TRAF2, a key element in the control of cell survival and apoptosis [31]. Involvement in the metabolism of lipids may be one more important biological mechanism of the AAM- and ANM-related role of TNFRSF11A. The relationship between obesity and AAM/ANM has been well documented [32, 33]. Arachidonic acid/prostaglandin E2 axis was implicated in uterine epithelium cell death induced by menopause [34]. The fatty acid composition was shown to be related to the menopausal status [35].
The lack of the GO Ontology data about RELCH/KIAA1468, PIGN, and LINC02341 may suggest that their role in metabolic pathways is still poorly studied. On the other hand, there is extensive evidence about co-expression of RELCH/KIAA1468 and PIGN with many genes, including those involved in the control of the basic cellular processes, e.g., cell proliferation [36] (Supplementary Table 4).
In general, a degree of gene pleiotropy seems to be inversely related to the relative contribution of the gene to the trait. Given that most genes in the human genome are pleiotropic [37], the expected contribution of each of them to a particular trait is quite modest. Therefore, highly pleiotropic genes have a small effect size and often yield false negative results in GWAS unless their contribution to a particular trait is above the average for other traits (e.g., TNFRSF11A/TNFSF11 contribution to bone remodeling).
The present study also sheds light on the frequently observed inconsistencies in associated polymorphisms and unsuccessful attempts to replicate candidate loci in different ethnic populations. Previous studies suggested that differences in population genetic structure might underlie the above disparities [38, 39]. The results of the present study suggest that, in addition to the allele frequencies, population-specific LD patterns are another important factor.
Conclusion. The in silico analysis of the TNFRSF11A and TNFSF11 polymorphisms previously reported for association with AAM and/or ANM suggested RELCH/KIAA1468, LINC02341, and AKAP11genes as candidates for the traits. While this assumption is biologically plausible, candidate gene association studies are needed to verify it. In summary, the present study demonstrates that the in-depth analysis of rapidly expanding biological databases may provide new insights into possible factors and mechanisms underlying the observed association of genetic markers with a trait.













Reference lists