
A novel heterozygous variant in exon 32 of the CHD7 gene (c.6923C>T) in a Syrian family with Kallmann syndrome
Aннотация
Background: Kallmann syndrome (KS) and CHARGE syndrome (CS) are rare heritable disorders in which anosmia and hypogonadotropic hypogonadism co-occur. KS is genetically heterogeneous with at least eight genes being involved in its pathogenesis, whereas CS is caused by autosomal dominant mutations exclusively in CHD7 gene. The majority of CS-cases are sporadic and only few familial cases have been reported. In these families, mosaicism in one parent, as well as parent-to-child transmission of a CHD7 mutation, were described. The aim of the study: To report a paternal transmission of a variant in exon 32 of the CHD7 gene (c.6923C>T) in a familial case originally suggested to be affected by KS. Materials and methods: Five genes associated with KS were analyzed using Sanger sequencing and MLPA in a 17-year-old male. Results: The heterozygous variant leading to a change of amino-acid serine at position 2,308 to leucine was found in father his three children. Conclusion: Overall this report confirms the existence of KS without CS symptoms, caused by a mutation in a gene reported pathogenic only in CS.
К сожалению, текст статьи доступен только на Английском
Introduction. Idiopathic hypogonadotropic hypogonadism (IHH; OMIM 146110), one of the most commonly inherited forms of diminished functional activity of the gonads, results from deficient hypothalamic of gonadotropin releasing hormone (GnRH) release or action [1]. IHH patients present with absent or impaired sexual development due to sex-steroid-hormone deficiency, low serum levels of the pituitary gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and infertility [1]. Kallmann syndrome (KS; OMIM 147950) is a combination of congenital hypogonadotropic hypogonadism (HH; OMIM 146110) and decreased/absent sense of smell [2]. Anosmia, or the inability to smell, is the result of olfactory bulb defects [3], whereas HH presents as absent or impaired pubertal maturation and is caused by GnRH deficiency [4]. KS accounts for about 50% to 60% of IHH [5] while forms of IHH present with normal olfaction (i.e. norm-osmic idiopathic HH: nIHH). KS is clinically and genetically very heterogeneous; phenotypic features may include additionally cleft lip/palate, hearing impairment, dental agenesis, limb anomalies, renal agenesis, and mirror movements [6]. The majority of KS cases (~60%) are sporadic; i.e. only one person is affected in the family. In familial KS, autosomal recessive, autosomal dominant, and X-chromosomal recessive inheritance have been described [7]. Oligogenic mode of inheritance has also been suggested [8, 9]. At present, mutations in eight genes explain approximately 25–35% of KS cases. Heterozygous loss-of-function mutations in the CHD7 gene were identified in patients with nIHH, KS, and CHARGE syndrome (CS) [10, 11]. CS is a highly variable disorder in which congenital anomalies, multisensory impairment, and variable mental retardation can occur (OMIM 214800). CHARGE is an acronym for ocular coloboma, heart defects, choanal atresia, retardation of growth and/or development, genital hypoplasia and ear anomalies combined with deafness [12]. HH and anosmia are present in the majority of patients with CS [13]. Recently, it was proven that HH and anosmia may co-occur in CS, too [14], which means that KS can be considered as part of the phenotypic spectrum of CS. CHD7 mutations are found in more than 90% of patients with typical CS [15].
Here we report a family with one patient with symptoms resembling KS however lacking CS symptoms, and with a novel heterozygous variant in exon 32 of the CHD7 gene (c.6923C>T) resulting in an amino acid exchange (p.Ser2308Leu).
Material and Methods
Clinical information
A 17-year-old male was the fifth child born to consanguineous Syrian healthy parents with a remarkable family history (delayed puberty in the children). At birth of the index patient his mother and father were 34 and 36 years old, respectively. The mother reported no history of infection during this pregnancy except for a slight hemorrhage at its beginning. After otherwise uncomplicated pregnancy and delivery, the index presented with micropenis and small testes (Tanner scale I, data not shown). At 17 years medical analyses showed an FSH (follicle stimulating hormone) level of 0.15 (normal value: 1.5 – 12.4 mlU/ml), a LH (luteinizing hormone) level of 0.10 (normal value: 1.4 – 8.6 U/ml) and a testosterone total level of 1.9 (normal value: 2-8 nmol/l). The patient had a normal male 46,XY karyotype and azoospermia without AZF-chromosome microdeletions (results are not shown). Subsequent normal height (170 cm) and weight (68 kg), no coloboma, no choanal atresia, and no cardiovascular malformations. The patient suffered from anxiety, was nervous and slightly autistic. The patient’s older brother and sister (the second and third child of the family) also expired delayed puberty but disappeared spontaneously in the boy and in the girl after hormonal treatment for 1.5-2 years. The remainder family members were clinically healthy. The study was approved by the ethical committee of the Atomic Energy Commission, Damascus, Syria. Informed consents were obtained from the father and other family members.
Genetic analysis
Genomic DNA was extracted from peripheral blood using QIAamp DNA Blood Mini Kit (Qiagen GMBH, Hilden, Germany). Sequencing of the following genes was performed: Kallmann 1 KAL1 (ANOS; NM_000216), Kallmann 2 FGFR1 (NM_23110.2), Kallmann 3 PROKR2 (NM_144773), Kallmann 4 PROK2 (NM_001126128), Kallmann 5 CHD7 (NM_017780). Analysis was performed stepwise by Sanger-sequencing and analyses of deletions and duplications by Multiplex Ligation-dependent Probe Amplification (MLPA, MRC-Holland).
Results. No mutations, deletions or duplications were found in KAL1 (ANOS; NM_000216), FGFR1 (NM_23110.2), PROKR2 (NM_144773) or PROK2 (NM_001126128). However, in CHD7 (NM_017780) a novel heterozygous variant in exon 32 of the CHD7 gene (c.6923C>T) resulting in an amino acid exchange (p.Ser2308Leu) was identified (Fig. 1) in patient, as well as in older 22 year old sister, 24 year old brother and the father (Tab. 1).
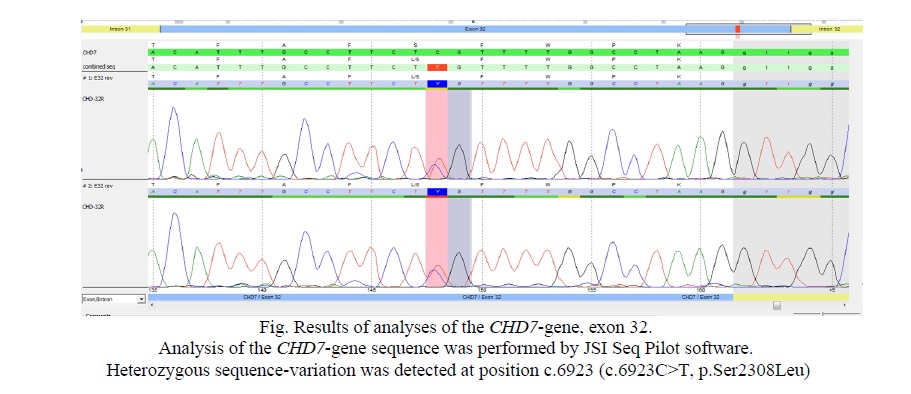

Discussion. KS is a unique form of IHH disease spectrum characterized by developmental disorders with olfactory abnormalities being caused by congenital defects in GnRH secretion of varying degrees. The pulsatile secretion of GnRH is essential for the hypothalamic pituitary-gonadal axis function [16]. Also, KS is a unique disease model to study the migration of GnRH neurons and the development of human puberty. Some genes are necessary for the correct differentiation, migration, upstream signal regulation, and function of GnRH neurons in the embryonic period, which can lead to IHH [16]. Some genes that correctly differentiate embryonic GnRH neurons may be correlated with IHH, such as KAL-1, FGFR1, FGF8, PROKR2, PROK2, CHD7, NELF, WDR11, HS6ST1, KISS1R, KISS1, TAC3, TACR3, LEPR, LEP, PCSK1, GNRHR, GNRH1, SEMA3A, and NDN7 [16]. Mutations in these genes lead to certain degrees of clinical manifestations. Moreover, KS can be caused by genes such as KAL-1, FGFR1, PROKR2, PROK2, CHD7, and FGF8 [16]. Among them, the CHD7 mutation was only found in KS patients with the CS phenotype, suggesting that if a patient was diagnosed with hypogonadism and anosmia, attention should be paid to the investigation of the presence of clinical characteristics of CS [16]. Here we report a paternally inherited new CHD7 mutation in a KS patient.
The CHD7 gene is located on chromosome 8q12.1, which encodes DNA-binding protein 7 of helicase in the chromatin region. This protein family has a unique functional domain binding site, including 2 N-terminal chromatin domains, 1 SWI2/SNF2-like ATP enzyme/solution helix domain, and 1 DNA binding domain. CHD7 protein complex is expressed in the olfactory epithelium, hypothalamus, as well as the pituitary gland, suggesting that this protein may play an important role in the development of the olfactory bulb and GnRH neuron migration. The genetic pattern of CHD7 gene has not yet been fully understood, and may follow autosomal dominant inheritance, with its mutations accounting for 6% of all IHH patients [17].
Jongmans et al. [10] identified 3/38 KS patients harboring de novo CHD7 mutations (2 stop-mutations and 1 missense) whereas the nIHH patients were negative for CHD7 variants. However, all 3 KS patients with CHD7 variants, upon additional phenotypic review, universally exhibited major CS features. In contrast, Kim et al. [11] identified 7/56 IHH patients (3KS, 4 nIHH) harboring CHD7 mutations (2 intronic mutations leading to exon skipping and 5 missense mutations), all of whom lacked major CS phenotypes, thus implicating CHD7 allelic variants as a cause of both KS and nIHH forms of IHH without CS features. In view of these conflicting data as to whether CHD7 mutations are capable of causing KS or nIHH without full CS, Bergman et al. [18] examined 36 KS patients in whom they identified 3 with CHD7 mutations (2 nonsense, and 1 de novo missense). However, all three subjects displayed additional CS features, leading to their conclusion that CHD7 mutations do not cause isolated IHH. Laitinen et al. [19] revealed no CHD7 mutations in 30 Finnish KS patients. Jie et al. [16] found a family (two sons inherited a mutation from his mother, but the mother and his younger son did not show clinical features of KS) suffering from KS and some clinical features of CS. Pedigree verification can be achieved by CHD7 gene mutation c.6571G>A. Hyung-Goo Kim et al. [11] examined 7/111 IHH/KS patients in whom they identified 7 CHD7 mutations (two splice and five missense), three unrelated probands with KS and four unrelated probands with IHH with CHD7 mutations, demonstrating that CHD7 is involved in either IHH or KS. However, Hyung-Goo Kim et al. [11] suggested new evidence for a role of CHD7 in the pathophysiology of both normosmic IHH and KS patients without a CS phenotype.
The molecular basis for 70%–75% of IHH/KS patients remains unknown [11]. To date, only FGFR1 mutations have been reported to cause either nIHH families or KS families. Although a homozygous PROKR2 deletion was seen in a single family comprising both normosmic and anosmic patients, this represents variable expressivity within the same family [11]. Interestingly, Hyung-Goo Kim et al. [11] found a one IHH and one KS patient, who both lack the CS phenotype, possess the same mutations (Ser834Phe and IVS65G/C) reported previously in patients with CS, further demonstrating the allelic relationship of both syndromes. The KS patient with the IVS65G/C mutation does not fulfill Blake’s criteria for CS, although she does have hearing impairment and cleft lip and palate. This also indicates that the effects of modifying genes may determine whether the patient has the more severe CS phenotype rather than the milder IHH/KS phenotype [11].
Overall, the family report confirms the existence of autosomal dominant inheritance of KS with lack of CS symptoms and a mutation in the yet only reported pathogenic gene involved in CS. This heterozygous variant in exon 32 of the CHD7 gene (c.6923C>T), leading to a change of amino acid serine at position 2,308 to leucine devoid of any known functional domain, and therefore, it is unlikely to be a dominant negative form of the protein. Therefore, we hypothesize that this mutation represents a null allele, causing disease due to haploinsufficiency. This is in agreement with previous findings suggesting that a full genetic dosage is required for complete function of CHD7 [18]. This novel variant in the present case was inherited from his father. Also, this variant was found in two subsequent children of his family. Moreover, this observation would draw the attention of the clinicians on the germline and familial responsible for the variable intrafamilial expression, suggesting a careful genetic counseling.













Список литературы
Список использованной литературы появится позже.