
Рак яичников в составе наследственных онкологических синдромов (обзор)
Aннотация
Актуальность: Наследственные формы рака яичников (РЯ) составляют более одной пятой случаев злокачественных новообразований данной локализации. Открытие новых молекулярно-генетических предикторов развития РЯ привело к совершенствованию системы ранней диагностики и терапевтических подходов к лечению, что позволило значительно сократить смертность от данной онкопатологии. Однако существующие системы скрининга охватывают лишь небольшой спектр патогенных вариантов, из-за чего их прогностическая значимость сильно снижается. Цель исследования:На основании изучения данных современной литературы рассмотреть представленность рака яичников в составе наследственных сидромов и оценить вклад генетических факторов в развитие наследственных форм рака яичников. Материалы и методы:Анализ литературных данных проводился по ключевым словам: наследственный рак яичников, синдром рака груди и яичников, синдром Коудена, синдром Линча, синдром Неймегена, атаксия-телеангиэктазия, анемия Фанкони, синдром Пейтца-Егерса за период 1981-2021 гг. в базах данных PubMed, PMC, eLibrary. Результаты:Синдром рака молочной железы и яичников является наиболее распространенной формой семейного РЯ, который в 65–85% случаев обусловлен герминальными мутациями в генах BRCA1/BRCA2. Однако на сегодняшний день известно еще по крайней мере шесть опухолевых синдромов, связанных с наследственным РЯ и обусловленных мутациями в других генах-супрессорах и онкогенах, включая гены MSH6,MLH1,MSH2 (синдром Линча), NBN (синдом Неймегена), АТМ (атаксия-телеангиэктазия), STK11 (синдром Пейтца-Егерса), RAD51С, RAD51D, BRIP1, PALB (анемия Фанкони), PTEN (синдром Коудена). В совокупности герминальные мутации в вышеупомянутых генах ответственны примерно за 15-20% случаев наследственных форм РЯ. Тем не менее спектр патогенных вариантов в этих генах и их вклад в развитие РЯ изучен недостаточно, что усложняет разработку молекулярных диагностических стратегий. Заключение:Разработка и внедрение новейших технологий секвенирования позволили существенно расширить знания о молекулярных механизмах опухолеобразования яичников и выявить множество новых молекулярных маркеров этого процесса. Однако вклад выявленных вариантов в формирование предрасположенности к РЯ изучен недостаточно и требует проведения дальнейших исследований.
Ключевые слова: наследственный рак яичников, синдром рака молочной железы и яичников, синдром Коудена, синдром Линча, синдром Неймегена, атаксия-телеангиэктазия, анемия Фанкони, синдром Пейтца-Егерса
Введение. Среди новообразований женской репродуктивной системы рак яичников является наиболее сложной формой онкопатологии, этиология и патогенез которой окончательно не изучены. Отсутствие патогномоничных начальных симптомов, диагностика рака на поздних стадиях, агрессивное клиническое течение, высокая смертность, несмотря на оптимизацию методов лечения, диктуют необходимость дальнейшего исследования данной проблемы. Рак яичников составляет 4-6% в структуре онкологической заболеваемости женщин, является седьмым по распространенности раком и восьмой ведущей причиной смертности от рака у женщин во всем мире [1]. Российская Федерация занимает лидирующие позиции по показателям заболеваемости раком яичников (10,2 случаев на 100 тыс. женщин в год). В России злокачественные опухоли яичников ежегодно выявляются более чем у 13 000 женщин, и около 8 000 женщин умирают от этого заболевания (рис. 1) [2].

По показателям смертности РЯ занимает первые строчки среди всех гинекологических опухолей в большинстве индустриальных стран мира, поскольку у большинства больных заболевание выявляется на поздних стадиях, когда общая пятилетняя выживаемость не превышает 30-40%. Летальность больных раком яичников на первом году после установления диагноза составляет 35% [3]. Лишь у 15% женщин заболевание обнаруживается на 1 стадии. Примечательным является тот факт, что показатели общей пятилетней выживаемости при РЯ практически не изменились с 1995 года, что свидетельствует об актуальности проблемы ранней диагностики данной онкопатологии [1].
Рак яичников – гетерогенное заболевание как с точки зрения этиологии, так и со стороны клинических проявлений. В основе происхождения всех опухолей данной локализации лежат мутации генетического аппарата клеток, которые формируют их повышенную чувствительность к воздействию экзогенных и эндогенных факторов. Такими факторами являются возраст, отсутствие беременностей и родов, приводящее к "непрекращающейся овуляции", применение гормональных препаратов, стимулирующих овуляцию, неправильное питание, вредные привычки и другие [4].
На сегодняшний день роль генетического фактора в этиологии рака яичников не вызывает сомнения. Результаты проведѐнных исследований показали, что риск развития заболевания для женщин с семейной историей РЯ повышается в 3-4 раза по сравнению с общей популяцией [5]. Наследственные формы РЯ составляют более одной пятой (около 23%) случаев злокачественных новообразований яичников [6]. В настоящее время идентифицировано, по крайней мере, шесть наследственных синдромов, проявляющихся семейной предрасположенностью к возникновению рака органов женской репродуктивной системы [7]. Однако наиболее изученными из них являются две независимые формы наследственного рака яичников: синдром рака молочной железы и яичников (СРМЖиЯ) и синдром Линча (СЛ).
Синдром рака молочной железы и яичников
СРМЖиЯ является наиболее распространенной формой наследственного РЯ (65-85% всех случаев). В подавляющем большинстве случаев синдром обусловлен герминальными мутациями в генах BRCA1 или BRCA2 [6]. Герминальные мутации BRCA1 и BRCA2 встречаются примерно у 20–30% пациенток с наследственным раком яичников [8, 9]. Вероятность развития заболевания у женщин с патогенными изменениями в гене BRCA1 возрастает до 20-50%, а с мутациями в гене BRCA2 до 5-23 %, по сравнению с показателями в общей популяции (риск развития РЯ в течении жизни – 1,6%) [10].
Многочисленные исследования показывают, что мутации в BRCA1/2 приводят не только к высокому риску развития РЯ в течение жизни, но и накладывают особенности на его клиническое течение. Для наследственного BRCA-ассоциированного РЯ характерен более молодой в сравнении со спорадическим РЯ возраст манифестации заболевания. В основном BRCA-позитивный РЯ характеризуется серозным гистологическим типом с высокой степенью злокачественности, а также высокой частотой ответа на первую и последующие линии платиносодержащей химиотерапии, длительными безрецидивными периодами и лучшей общей выживаемостью [11].
Ген BRCA1 (был выделен в 1994 году и картирован на хромосоме 17q21 [12]. Спустя год в области хромосомы 13q12–13 был обнаружен ген BRCA2 [13]. К настоящему времени получена существенная информация о структуре и функции этих генов. В норме они осуществляют контроль целостности генома. Утрата функции белков BRCA1/2 влечет за собой ошибки репарации двунитевых разрывов ДНК, вследствие чего инактивируются гены контроля клеточного цикла, ингибирующие дальнейший клеточный рост и индуцирующие апоптоз. Накопление ошибок репарации, которые приводят к нарушениям регуляции клеточного цикла, апоптоза и дифференцировки клетки, ведут к генетической нестабильности, что является ключевым событием в процессе злокачественной трансформации клетки [14].
На сегодняшний день идентифицировано более 3000 различных мутаций в генах BRCA1/2 [15]. В крупном популяционном исследовании, в котором принимали участие 29700 семей с мутациями BRCA1/2, было выявлено 1 650 уникальных вариантов в гене BRCA1 и 1731 уникальных вариантов в гене BRCA2. Большинство из них были представлены мутациями, приводящими к сдвигу рамки считывания, а также нонсенс мутациями, которые являляются причиной преждевременного прекращения трансляции и формирования нефункционального белка. Геномные перестройки и миссенс мутации составляют гораздо более высокую долю изменений в гене BRCA1 по сравнению с геном BRCA2, что, по мнению авторов, является причиной неравномерного распределения частот различных типов мутаций в разных популяционных группах [16]. Частота мутаций генов BRCA1/2 в общей популяций больных семейными формами РМЖ/РЯ оценивается от 1:300 до 1:800 в различных этнических группах [17].
При изучении генетической структуры РЯ в различных популяциях ученые отметили, что не только частота, но и спектр патогенных мутаций различается среди разных групп населения. В некоторых этнических группах представлен широкий спектр различных мутаций с низкой частотой, в то время как в других преобладают лишь несколько повторяющихся специфических мутаций. Такой феномен получил название эффекта основателя. На сегодняшний день мутации основателей в генах BRCA1/2 описаны у евреев Ашкенази, в польской, норвежской, исландской, славянской и других популяциях [18-26].Так, в популяции евреев Ашкенази наиболее частыми мутациями являются 185delAG (1%), 5382insC (0,1-0,15%) в гене BRCA1 и 6174delT (1,52%) в гене BRCA2. На их долю приходится до 30 % всех случаев заболеваемости наследственными формами РМЖ и РЯ [18]. Многие учёные также отмечают высокую частоту мутаций 5382insC и 185delAG в странах восточной Европы, включая Россию [27-29]. Было идентифицировано и несколько других этноспецифических мутаций, включая исландскую мутацию-основателя c.771_775del (999del5) в гене BRCA2 [19]; французско-канадские мутации c.4327C> T (C4446T)/BRCA1 и c.8537_8538del (8765delAG)/BRCA2 [20, 21]; мутации c.181T> G/BRCA1 и c.4034delA/BRCA1 в Центральной и Восточной Европе [22, 23]; c.548-4185del гена BRCA1 в Мексике [24], мутацию c.9097dup гена BRCA2 в Венгрии [25, 26] и другие.
Некоторые из таких мутаций имеют высокую распространенность и в других популяциях. Согласно недавнему популяционному исследованию, проводимому консорциумом CINBA и объединяющему данные из 49 стран по всему миру, наиболее распространенными мутациями оказались мутации с эффектом основателя еврейского происхождения c.5266dup (5382insC)/BRCA1, c.68_69del (185delAG)/BRCA1 и c.5946del (6174delT)/BRCA2. Так, мутация 5382insC с высокой частотой была выявлена в ряде европейских стран, таких как Россия, Польша, Чехия и Литва, где на нее приходится соответственно 94%, 60%, 33% и 50% всех мутаций в гене BRCA1. Еще одним примером служит мутация c.181T>G в гене BRCA1, предположительно имеющая восточно-европейское происхождения. Данная мутация наблюдалась в Центральной Европе (Австрия, Чехия, Германия, Венгрия, Италия и Польша) [16, 22, 23].
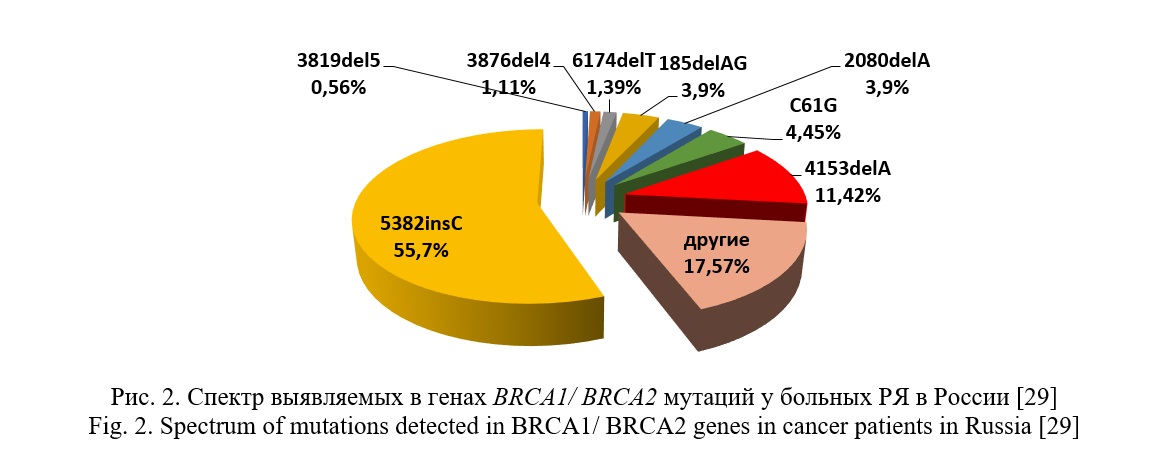
Согласно ряду российских исследований, преобладающими мутациями в генах BRCA1/2 на территории Российской Федерации являются: 5382insC, С61G, 185delAG, 4153delA, 2080delA, 185delAG, 3819delGTAAA, которые охватывают до 70–90 % всего спектра выявленных мутаций в этих генах [27-33] (рис. 2). Однако в связи с тем, что население Российской Федерации имеет сложный этнический состав, а также относительно изолированное существование некоторых популяций, спектр и частота мутаций в генах BRCA1 и BRCA2 разнится от региона к региону [27, 30, 31]. Так, в результате скрининга мажорная мутация 5382insC была обнаружена лишь у 7% женщин татарской этнической принадлежности, тогда как у женщин со славянским происхождением данная мутация встречалась в 5 раз чаще [32].
Значимые патогенные варианты в гене BRCA2 у больных РЯ в российской популяции встречаются редко. По данным российских исследований их частота составляет примерно 1,4-2% [8, 9, 27-29]. Для данного гена характерно отсутствие «горячих точек» и высокая доля (30%) вновь выявленных мутаций, что определяет необходимость скрининга всей кодирующей части данного гена [33].
Интересным направлением исследования является поиск локусов, которые могут снижать или увеличивать риск развития РЯ у носителей мутаций в BRCA1/2. Так, в результате полногеномного ассоциативного исследования (GWAS) было выявлено несколько однонуклеотидных полиморфных локусов (SNP), связанных с повышенным риском развития рака яичников у женщин в общей популяции [34]. Четыре из этих SNP (rs10088218, rs2665390, rs717852, rs9303542) были ассоциированы с повышенным риском РЯ у носителей патогенных вариантов в гене BRCA2, тогда как локусы rs10088218 и rs2665390 были связаны с более высоким риском развития заболевания у носителей мутаций в гене BRCA1 [35]. Некоторые из таких генетических маркеров могут быть ассоциированы с определенным гистотипом опухоли. Так Kuchenbaecker с коллегами показали, что сочетанное носительство генетических вариантов 1p36 (WNT4), 4q26 (SYNPO2), 9q34.2 (ABO) и 17q11.2 (ATAD5) с мутациями в генах BRCA1/2 увеличивают риск всех подтипов эпителиального РЯ, в то время как мутации 1q34.3 (RSPO1) и 6p22.1 (GPX6) увеличивают риск серозного рака яичников у носителей патогенных вариантов в генах BRCA1/2 [36]. Таким образом, изучение патогенных вариантов BRCA1/2 в комплексе с другими генетическими вариантами могут способствовать более точному прогнозу риска развития РЯ.
Синдром Линча
В середине 1960-х годов Линч и его коллеги описали аутосомно-доминантный наследственный синдром, который предрасполагал молодых людей (средний возраст 45 лет), не пораженных аденоматозными полипами толстой кишки, к развитию колоректального рака [37]. В последующих публикациях по этому синдрому сообщалось, что члены этих семей также были склонны к избыткам экстраколонических раков, включая рак эндометрия, яичников, желудка, тонкой кишки, гепатобилиарного тракта, поджелудочной железы, почечной лоханки, мочеточника, молочной железы, простаты и головного мозга [38, 39]. Заболеваемость синдромом Линча (СЛ) оценивается от 1:370 до 1:2000 в западных популяциях [38].
Этиологической причиной развития СЛ являются герминальные мутации в генах, участвующих в многоступенчатом механизме репарации неправильно спаренных оснований ДНК, известном как мисмэтч репарация (MMR). Белки MSH2 и MSH6 формируют гетеродимер, функцией которого является выявление некомплементарных оснований, а также инсерций и делеций, которые могут возникать в процессе репликации ДНК. При обнаружении ошибки к данному комплексу присоединяются белки MLH1, PMS1 или PMS2, которые участвует в восстановлении нити ДНК [40].
Инактивация генов системы MMR приводит к накоплению повторяющихся нуклеотидных последовательностей, вызывая состояние, называемое микросателлитной нестабильностью. Микросателлиты представляют собой короткие тандемные повторяющиеся последовательности ДНК с высокой восприимчивостью к ошибкам репликации. Такие участки содержат некоторые онкогены и гены опухолевых супрессоров, а также гены репарации двунитевых разрывов ДНК, следовательно, дефекты системы MMR могут опосредованно приводить к нарушению работы генов, поддерживающих стабильность генома, таким образом запуская процесс канцерогенеза [41].
СЛ является второй по частоте причиной наследственного эпителиального РЯ, составляя 10-15% [42]. В общей структуре эпителиального РЯ данный синдром составляет примерно 0,5-3% [43]. РЯ в 3 раза чаще встречается у женщин с СЛ по сравнению с общей популяцией. Дефицит системы MMR встречается приблизительно в 10-12% случаев эпителиального РЯ [44]. В частности, он обнаруживается в 19,2% эндометриоидных, 16,9% муцинозных, 11,5% светлоклеточных и 1–8% серозных гистологических подтипов. Распространенность MMR-дефицита или микросателлитной нестабильности (MSI) при семейном РЯ оценивается от 10% до 20% [45-47].
С клинической точки зрения, РЯ при СЛ характеризуется ранним началом заболевания (в среднем 41-49 лет) и в основном имеет несерозную гистологию с преобладанием эндометриодных, муцинозных и светлоклеточных гистологических типов. Кроме того, в 65–80% случаев РЯ, связанный с СЛ, диагностируется на ранних стадиях и по этой причине имеют более благоприятный прогноз выживаемости по сравнению со спорадическим или наследственным РЯ, вызванным мутациями в генах BRCA1/2 [48].
По последним данным предполагается, что кумулятивный риск развития рака яичников может достигать 10% для носителей мутаций в гене MLH1 в возрасте 75 лет, 17% для гена MSH2 и 13% для гена MSH6 [49]. Тогда как для гена PMS2 он составил менее 1% [50]. В ходе исследований «случай-контроль» было установлено, что риск развития РЯ в 3 раза выше у носителей мутаций в гене MLH1 [49], от 2 до 14 раз – у пациенток с изменениями в гене MSH2 [51-54], и от 2 до 9 раз – у больных с мутациями в гене MSH6 [51-55]. При этом сочетанное носительство комбинации патогенных вариантов генов системы MMR приводило к 2-кратному увеличению риска развития РЯ, а кумулятивный риск рака яичников к возрасту 80 лет составил 3,7% [56].
Распространенность патогенных герминальных вариантов в генах MMR среди пациенток с диагнозом РЯ невысока и оценивается в 0,5-3%, что объясняется их связью с редкими гистотипами при данном заболевании [51-57]. По этой причине некоторые ученые считают целесообразным при подозрении на СЛ пациенткам с РЯ предварительно проводить скрининг на наличие микросаттелитной нестабильности [56].
Наиболее подробно спектр мутаций в генах MMR описан в работе Pal T. В результате генетического тестирования кодирующих областей генов MLH1, MSH2 и MSH6 у 1893 пациенток с диагнозом «рак яичников» было выявлено 161 изменение нуклеотидной последовательности. Из всех идентифицированных изменений лишь 9 вариантов были классифицированы как патогенные. Два из них (c.676C>T и c.1852_1854delAAG) локализованы в гене MLH1, два (c.163delC и c.2038C>T) в гене MSH2 и пять (c.1636 G>T c.2150_2153delTCAG c.2690_2691insA c.2731C>T c.3103C>T) в гене MSH6. Все выявленные патогенные изменения были представлены в единичных случаях [58].
Атаксия-Телеангиэктазия
Ген ATM (ATM serine/threonine kinase) расположен в локусе 11q22-23 и кодирует серин / треониновую протеинкиназу, играющую центральную роль в качестве активатора каскада реакций в ответ повреждение ДНК после двухцепочечных разрывов ДНК и функционирующую как регулятор широкого спектра белков, включая белки-супрессоры опухолевого роста P53, CHEK2 и BRCA1 [59].
Герминальные мутации в гене ATM являются причиной Атаксии-Телеангиэктазии (АТ), редкого аутосомно-рецессивного синдрома, проявляющегося множеством фенотипических характеристик, включая нейродегенерацию, мозжечковую атаксию, иммунодефицит, дисгенезию гонад, радиочувствительность и повышенный риск развития злокачественных новообразований [60].
Согласно M.Swift, среди родственников больных АТ моложе 45 лет смертность от онкологических заболеваний в 5 раз выше по сравнению с общей популяцией. Кроме того, в таких семьях отмечался высокий уровень заболеваемости карциномой яичников, желудка, молочной железы, желчевыводящих путей и другими неоплазиями [61].
Патогенные варианты в гене АТМ распознаются в 0,64-2,3% случаев наследственного РЯ [62-64]. В ряду исследований «случай-контроль» было установлено, что патогенные варианты в гене ATM могут быть связаны с умеренным риском развития семейных случаев РЯ, примерно в 2 раза превышающим популяционный [65-68]. Важно отметить, что связь патогенных вариантов гена АТМ с развитием РЯ не зависит от наличия в личном или семейном анамнезе пациенток случаев заболевания РМЖ [65]. В настоящее время идентифицировано более 300 изменений в нуклеотидной последовательности гена ATM, большую часть которых составляют нонсенс мутации и мутации сайта сплайсинга [64]. Однако спектр изменений у больных РЯ изучен недостаточно.
В результате скрининга 333 пациенток с РЯ из Польши у одной был выявлен патогенный вариант c.6095G> A ATM, приводящий к нарушению процесса сплайсинга и утрате 43 экзона гена ATM. У женщины был диагностирован серозный РЯ [65]. Эта же мутация была ранее обнаружена у пациентки, с отягощенным анамнезом РМЖ и РЯ из Австрии [66], а также в семьях с синдромом АТ из Польши [67].
Семь патогенных мутаций в гене АТМ было выявлено в 10 семьях с наследственными формами РМЖ/РЯ из Австрии. Пять из них 687delA/ATM, 1802G>T/ATM, 2465 T>G/ATM, 6095 G>A/ATM и IVS10-6 T>G/ATM представляли собой мутации, приводящие к укорочению полипептидной цепи, два других изменения 8734A>G/ATM и 9031A>G/ATM - миссенс-мутации, предположительно влияющие на киназную функцию белка. Миссенс вариант 8734A>G/ATM наблюдался в двух разных семьях, соответствующих критериям наследственного РМЖ/РЯ. В одной из семей были зарегистрированы случаи двустороннего РМЖ и РЯ. У носительницы патогенного варианта 1802G>T, приводящего к утрате 13 экзона гена АТМ, был диагностирован РЯ в молодом возрасте. В семейном анамнезе были выявлены случаи заболевания раком мозга, печени и кожи. Наиболее частая обнаруживаемая мутация, IVS10-6T> G/ATM, была выявлена в трех семьях с наследственным РМЖ, а также у одной здоровой женщины без семейного анамнеза заболевания [66].
В нескольких исследованиях сообщалось о высокой частоте варианта c.7271T>G/ ATM среди больных семейными и спорадическими формами РМЖ [68, 69]. А в недавней работе Hall M.J. было установлено, что данный патогенный вариант также связан с умеренным риском развития РЯ [70].
Синдром Неймегена
Первый клинический случай синдрома Неймегена (СН) был описан в 1981 году учеными из Университета Неймегена в Нидерландах и изначально получил название синдрома хромосомных поломок Неймеген [71]. Данное заболевание относится к группе врожденных синдромов с хромосомной нестабильностью, куда также относятся анемия Фанкони, синдром Блума, атаксия-тельангиэктазия и пигментная ксеродерма. СН имеет ряд характерных для этой группы заболеваний особенностей, включая специфические хромосомные перестройки, комбинированный первичный иммунодефицит, чувствительность к ионизирующему излучению и повышенный риск развития злокачественных новообразований [72].
Молекулярной основой развития заболевания являются двуаллельные мутации в гене NBN. Ген был картирован в 1998 году на длинном плече восьмой хромосомы (8q21) и первоначально имел название NBS1 [73]. Белковый продукт этого гена является частью тримерного ядерного комплекса MRN (MRE11-RAD50-NBN), являющегося ключевым участником практически всех этапов репарации двуцепочечных разрывов от распознавания повреждений в цепи ДНК и запуска АТМ-сигнального каскада до восстановления структуры молекулы [74]. Нарушение работы комплекса MRN может приводить к накоплению многочисленных повреждений ДНК в клетках, и, как следствие, к их малигнизации. По этой причине у пациентов с СН и со схожими состояниями отмечается повышенная частота возникновения злокачественных новообразований различных локализаций, включая РЯ и РМЖ [75].
По данным нескольких популяционных исследований герминальные мутации в гене NBN встречаются примерно у 0,28-1% пациенток с РЯ [49, 63, 76, 77], однако их роль в развитии заболевания до сих пор остается предметом дискуссий. В нескольких исследованиях сообщалось об умеренном риске развития РЯ для носителей патогенных вариантов в гене NBN [53, 62, 75]. Тогда как ряд других исследователей не подтвердили данную связь [49, 74].
Одной из наиболее изученных и часто обнаруживаемых мутаций в гене NBN является делеция с.657_661del5/NBN. Частота носительства этого варианта может достигать 1,5% в странах Восточной Европы (Польше, России, Украине и др.), что связано с «эффектом основателя» [78, 79]. Данная мутация вызывает сдвиг рамки считывания, что приводит к возникновению преждевременного стоп-кодона и как следствие к укороченному белковому продукту. Однако функциональные исследования показали, что два новых стартовых кодона, созданные сдвигом рамки считывания, могут генерировать усеченные фрагменты белка NBN, тем самым частично сохраняя его функциональность [80].
В нескольких исследованиях сообщалось о низкой частоте встречаемости мутации c. 657del5/NBN среди больных РЯ, сопоставимой с таковой в контрольной группе [81, 82]. Тогда как в работах польских и сербских ученых патогенный вариант был выявлен у пациенток с РЯ с частотой от 1,2% до 1,7% [65, 83, 84].
Другое наиболее часто обнаруживаемое изменение в гене NBN представляет собой миссенс вариант c.511A> G, приводящий к замене изолейцина на валин в 171 положении. Данный вариант широко распространен в европейской и азиатской популяциях и по некоторым данным может способствовать развитию злокачественных опухолей различной локализации. Так в литературе он был описан у лиц, страдающих РМЖ, РЯ, раком легких, раком головного мозга, колоректальным раком, а также различными формами лейкемии [65, 84, 85]. Роль варианта c.511A> G/NBN в патогенезе РМЖ и РЯ до сих пор неясна. Roznowski в своей работе продемонстрировал высокий риск развития РМЖ для носителей данного изменения [85]. Однако большинство ассоциативных исследований не выявило достоверной связи между наличием варианта c.511A> G/NBN и развитием наследственных и спорадических форм РМЖ и/или РЯ и на сегодняшний день он значится как вариант с неопределенной патогенностью [84, 86, 87]. Что касается генов MRE11 и RAD50, по-видимому, их вклад в развитие наследственных форм РЯ невелик. Несколько авторов сообщали о наличии вероятно патогенных вариантов у пациентов с семейными и спорадическими формами РЯ и РМЖ [88, 89]. Однако крупные популяционные и исследования типа «случай-контроль» не обнаружили связь мутаций в генах MRE11 и RAD50 с развитием РЯ [51, 53, 62]. Таким образом, на сегодняшний день роль генов MRE11 и RAD50 в патогенезе РЯ окончательно не установлена, что диктует необходимость проведения дальнейших функциональных и клинических исследований.
Синдром Коудена
В 1980-х годах в ходе цитогенетических и молекулярных исследований было установлено, что при различных типах злокачественных опухолей обнаруживалась частичная или полная утрата 10-й хромосомы [90]. Дальнейшие исследования в этом направлении привели к открытию нового гена-супрессора опухоли PTEN в 1997 году [91]. Ген PTEN кодирует фосфатидилинозитол-3,4,5-трифосфат-3-фосфатазу, способную дефосфорилировать фосфопептиды и фосфолипиды. Онкосупрессорная активность PTEN связана c его способностью дефосфорилировать липидный субстрат — фосфатидилинозитол-3,4,5-трифосфат (PIP3), что приводит к ингибированию передачи сигналов по PI3K/AKT/mTOR-сигнальному пути, являющегося основным путем роста и пролиферации клеток [92]. Таким образом, белковый продукт гена PTEN является одним из ключевых супрессоров опухолей в организме.
Герминальные патогенные варианты в гене PTEN связаны с синдромом Коудена, аутосомно-доминантным заболеванием, характеризующимся доброкачественными гамартомами, а также повышенным риском РМЖ, рака щитовидной железы, матки и других видов опухолей в течение жизни [93]. Кумулятивный риск РМЖ у носителей патогенных/условно патогенных вариантов в гене PTEN оценивается в 25-85%, а риск развития карциномы эндометрия матки составляет 28,2% [94], в то время как о повышенном риске карциномы яичников в случаях с патогенными вариантами в гене PTEN не сообщалось [95]. Однако считается, что эндометриоидная карцинома яичников развивается из ткани эндометрия при ретроградной менструации и имплантации в яичник [96].
Мутации в гене PTEN распознаются в 20% случаев эндометриоидных карцином яичников [97], а LOH составляет от 60 до 64%, тогда как эти значения намного ниже при других типах карцином яичников (2% частота мутаций; 28% LOH) [98, 99]. Эти данные предполагают специфическую ассоциацию изменений нуклеотидной последовательности гена PTEN и РЯ эндометриоидного типа. Ранее в литературе было описано несколько клинических случаев развития РЯ у носительниц герминальных патогенных вариантов в гене PTEN, приводящих к полной потери его экспрессии. При генетическом тестировании пациенток было выявлено три различных герминальных варианта: нонсенс мутация p.Q219X/PTEN, мутация сайта сплайсинга c.1026 + 1G > T/PTEN и нонсенс мутация c.388C>T/PTEN. Интересно отметить, что опухоли во всех трех случаях различались гистологически. У носительницы варианта p.Q219X/PTEN была диагностирована дисгерминома яичников [99], у пациентки с патогенным вариантом c.1026+1G >T/PTEN был обнаружен двусторонний эндометриоидный рак яичников [100], а пациентка с мутацией c.388C>T/PTEN страдала от светлоклеточной карциномы [101].
Синдром Пейтца-Егерса
Ген STK11 расположен на коротком плече хромосомы 19 и кодирует внутриклеточную серин-треониновую киназу, участвующую в клеточном энергетическом метаболизме, пролиферации и поляризации клеток, p53-зависимом апоптозе, а также в регуляции внутриклеточных сигнальных путей VEGF и Wnt [102]. Ген STK11 представляет собой опухолевый супрессор, который мутирует при различных спорадических формах рака. Герминальные мутации в этом гене приводят к развитию редкого аутосомно-доминантного заболевания Синдрома Пейтца-Егерса (СПЕ), характеризующегося гиперпигментации слизистых оболочек кожи, гамартомам по всему желудочно-кишечному тракту, а также повышенным риском многочисленных злокачественных новообразований, в том числе гинекологических. Так риск рака яичников, связанный с СПЕ, по разным оценкам составляет 18-21% [103, 104]. Кроме того, женщины с этим синдромом подвержены развитию опухолей полового канатика с кольцевыми канальцами яичника, которые в 36% случаев возникают в связи с этим синдромом. Гистологически они представляют собой неэпителиальные доброкачественные опухоли, обладающие низким риском злокачественной трансформации [105].
STK11 относится к высокопенетрантным генам [7]. В работе Allison W. Kurian ген STK11 был связан с 40-кратным увеличением риска развития РЯ (OR=41.9; 95% CI, 5.55 to 315) [49]. Частота герминальных мутаций среди пациентов с эпителиальным РЯ варьирует от 0,23 до 1,61% [49, 106, 107]. На сегодняшний день в гене STK11 описано более 400 мутаций, приводящих к развитию фенотипа СПЕ [108]. Точечные изменения встречаются на всей протяженности гена STK11 и часто представлены нонсенс мутациями или мутациями сдвига рамки считывания. Большинство из них, по прогнозам, являются патогенными [109]. Однако спектр дефектов у пациенток с РЯ охарактеризован недостаточно. В работе N.Resta патогенные мутации в гене STK11 были обнаружены у трех пациенток с СПЕ, страдающих РЯ. По функциональной роли обнаруженные варианты представляли собой нонсенс мутации (c.292G>A и c.498 C>G) и мутацию сайта сплайсинга (c. 290+2 T>A). У носительниц мутаций c.498C>G/STK11 и c. 290+2T>A/STK11 наряду с РЯ был диагностирован рак шейки матки и РМЖ, соответственно [103]. Патогенный вариант c.1276C>T/STK11 был выявлен у одной из 120 женщин с наследственным РМЖ/РЯ в Южной Корее [110]. В китайской популяции нонсенс мутация 658C>T/STK11 была обнаружена у одной пациентки в возрасте 31 год с СПЕ, страдающей РЯ [111].
Анемия Фанкони
Анемия Фанкони (АФ) является редким генетическим заболеванием, с преимущественно аутосомно-рецессивным типом наследования. Этиологическим фактором развития болезни являются герминальные мутации, возникающие в генах системы репарации ДНК, что приводит к широкому спектру клинических проявлений вариабельной пенетрантности, в основном характеризующихся симптомами прогрессирующей костномозговой недостаточности, врожденными дефектами и предрасположенностью к злокачественным новообразованиям [112].
Несмотря на то, что число людей, затронутых АФ при рождении, очень мало (1 на 160 000 человек во всем мире), частота моноаллельных носителей намного выше, так в Северной Америке она составляет 1:181 человек, а в Израиле – 1:93 [113]. Соматическая утрата второго аллеля значительно повышает риск развития онкологических заболеваний, включая РЯ [114].
На сегодняшний день описано по крайней мере 22 гена, ассоциированных с развитием АФ или клинически схожих состояний: FANCA, FANCB, FANCC, FANCD1 / BRCA2, FANCD2, FANCE, FANCF, FANCG / XRCC9, FANCI, FANCJ / BRIP1, FANCL / PHF9, FANCM, FANCN / PALB2, FANCP / SLX4, FANCQ / ERCC4, FANCR / RAD51, FANCS / BRCA1, FANCT / UBE2T, FANCU / XRCC2, FANCV / REV7 и FANCW / RFWD3 [115]. Белки, кодируемые этими генами, участвуют в репарации поперечных межхроматидных сшивок, включая процессы восстановления ДНК и поддержания стабильности генома, за что они получили название АФ-протеины, а сам процесс путь АФ. Кроме того, они участвуют в процессах гомологичной рекомбинации и негомологичного соединения концов [116].
Ранее в данном обзоре уже были описаны моноаллельные мутации в генах BRCA1/2, которые существенно повышают риск развития семейных форм РЯ. Дальнейшее изучение пути АФ, привело к открытию еще ряда генов, связанных с наследственной предрасположенностью к РМЖ и/или РЯ.
Ген PALB2
Еще более десяти лет назад появились первые работы о связи патогенных герминальных вариантов в гене PALB2 с повышенным риском развития РМЖ, что в дальнейшем было подтверждено многочисленными исследованиями. Обобщением всех предыдущих работ стало крупное международное исследование на основе данных из 524 семей, в котором предполагаемый абсолютный риск у носительниц герминальных патогенных вариантов в гене PALB2 в возрасте 80 лет составил 53% для РМЖ и 5% для РЯ [117].
В последние годы ген PALB2 привлекает внимание ученых в качестве гена предрасположенности к РЯ. Белок PALB2 является основным партнером BRCA2, который необходим для обеспечения его устойчивости и внутриядерной локализации, а также для привлечения его в сайты повреждения ДНК [118]. Другой важной функцией PALB2 служит образование «комплекса BRCA», в котором PALB2 служит молекулярным каркасом между BRCA1 и BRCA2 [119]. В свою очередь комплекс BRCA1-PALB2-BRCA2 необходим для рекрутирования белка RAD51 в место повреждения ДНК и инициации гомологичной рекомбинации. Таким образом, белковый продукт гена PALB2 имеет решающее значение для инициации гомологичной рекомбинации ДНК и играет ключевую роль в поддержании стабильности генома. Двуаллельные герминальные мутации в гене PALB2 приводят к анемии Фанкони (тип анемии Фанкони N), тогда как моноаллельные мутации связаны с повышенным риском рака молочной железы, поджелудочной железы и, возможно, яичников [117]. В работе L. Castéra было показано 8-кратное увеличение риска развития наследственного РМЖ и РЯ у носителей патогенных вариантов в гене PALB2. Однако значимых различий в частоте встречаемости мутаций в семьях с НРЯ и контролем обнаружено не было [52]. В то же время H. Song в своей работе продемонстрировал связь мутаций в гене PALB2 с развитием серозного РЯ высокой степени злокачественности [55].
Согласно популяционным исследованиям частота герминальных мутаций в гене PALB2 среди пациенток с семейными формами РЯ составляет 0,28-0,62% [57, 63, 120, 121]. Спектр мутаций в гене PALB2 аналогичен таковому в генах BRCA1 и BRCA2. Большинство изменений представлены точечными мутациями, подавляющую часть которых составляют миссенс-мутации, мутации сдвига рамки считывания и нонсенс мутации [53]. Однако, в отличие от партнеров, частота патогенных вариантов в гене PALB2 невысока, и у пациенток с раком яичников колеблется от 0,21% до 1% [51, 54, 57, 63, 75]. Для гена PALB2 выявлено несколько мутаций с эффектом основателя. Так мутация c.2323C> T/PALB2 была обнаружена у 0,4-0,7% больных РМЖ и у 0,6% больных РЯ франко-канадского происхождения в четырех независимых исследованиях [122, 123]. Интересно отметить, что данная мутация была обнаружена у пациенток преимущественно с ранним возрастом развития РМЖ.
Для жителей Центральной и Восточной Европы характеры две рекуррентные мутации в гене PALB2, связанные с РМЖ. Одна из них c.509_510delGA/ PALB2 – это европейская мутация-основатель, которая была обнаружена у 0,6-1,7% пациентов с РМЖ из Польши, в общей выборке больных РМЖ из Германии (0,3%), России (0,2%) и Белоруссии (0,3%) [124, 125].
Другая мутация, c.172_175delTTGT/PALB2 была идентифицирована у 0,3% пациентов в общей выборке РМЖ и 0,7% пациентов с семейными формами РМЖ из Польши [126], 0,9% пациентов в общей выборке больных РМЖ из Чехии [127], а также у 0,1% больных с семейными формами РМЖ из Германии [128] и 0,4% больных РЯ и 0,5% РМЖ из России [129]. Мутация c.509_510delGA/PALB2 способствовала развитию наследственных форм РМЖ/РЯ, но отсутствовала у пациентов в общей выборке РМЖ/РЯ, в то время как мутация c.172_175delTTGT/PALB2 была идентифицирована как у пациентов в общей выборке РМЖ/РЯ, так и в группе пациенток с отягощенным семейным анамнезом [130].
В польской популяции мутация c.509_510delGA/PALB2 достоверно чаще встречалась в группе больных РЯ (0,38%), по сравнению с контрольной группой (0,06%). Тогда как частота патогенного варианта c.172_175delTTGT оказалась примерно одинаковой в обеих группах [128].
Таким образом, можно сделать вывод, что вклад патогенных вариантов гена PALB2 в развитие РЯ выражен в значительно меньшей степени, по сравнению с генами BRCA1 и BRCA2. Тем не менее, имеющиеся литературные данные не позволяют сделать однозначный вывод об отсутствии или наличии повышенного риска развития РЯ у носителей мутаций в гене PALB2.
Гены RAD51C и RAD51D
Результаты нескольких недавних исследований показывают, что после BRCA1/2 гены RAD51C и RAD51D могут быть наиболее важными генами предрасположенности к НРЯ. В совокупности герминальные мутации в этих генах составляют ~ 1% случаев РЯ [53]. Интересно отметить, что в отличии от других генов-кандидатов наследственных форм РМЖ и РЯ, таких как BRCA1/2, TP53, PTEN, NBN, гены RAD51C и RAD51D, по-видимому, являются генами предрасположенности к наследственным формам РЯ, но не РМЖ [53, 65].
Белковые продукты генов RAD51C и RAD51D входят в семейство RAD51, которое также включает паралоги RAD51, RAD51B, XRCC2 и XRCC3. Белки RAD51 участвуют в процессе репарации двухцепочечных разрывов посредством гомологичной рекомбинации, функционируя как хранители генома. Избыточная экспрессия или потеря их функций приводит к геномной нестабильности [132].
Первое исследование гена RAD51C в качестве гена-кандидата РЯ и РМЖ было проведено А. Meindl с коллегами в 2010 году [133]. Тогда было показано, что герминальные патогенные мутации в этом гене, выявленные у 1,3% семей с наследственным синдромом РМЖ и РЯ, предрасполагают к РЯ, но не связаны с РМЖ. Спустя год в своей работе С. Loveday с коллегами показали, что герминальные мутации в гене RAD51D в шесть раз увеличивают риск развития РЯ в семьях с отягощенным анамнезом РМЖ и РЯ, тогда как связь с РМЖ оказалась статистически незначимой [134]. С тех пор роль паралогов RAD51C и RAD51D в качестве генов предрасположенности к НРЯ была неоднократно доказана в ряду исследованиях [121, 135, 136]. Вероятность развития РЯ у носителей патогенных изменений в генах RAD51C и RAD51D к 80 годам составляет 11% и 13%, соответственно [137]. Согласно данным метаанализа, проведенном M.Suszynska с коллегами на основе объединенных данных из 63 исследований, частота патогенных вариантов в генах RAD51C и RAD51D среди больных РЯ составила 0,62% и 0,41%, соответственно. При сравнении частот встречаемости патогенных вариантов среди пациенток с РЯ и контроля было установлено, что относительный риск развития заболевания был более чем в пять раз выше у носительниц мутаций в гене RAD51C и практически в 7 раз – у пациенток с патогенными изменениями в гене RAD51D [138].
Ранее L.Castéra с коллегами был проведен поиск патогенных вариантов в 34 генах-кандидатах РМЖ и РЯ в выборке, включающей более 5 тысяч женщин с наследственными формами РМЖ и/или РЯ. При этом частота патогенных вариантов в генах RAD51C и RAD51D составила, соответственно, 0,53% и 0,22%, а наличие этих изменений было ассоциировано с 4-кратным и 5-кратным повышением риска развития наследственных форм РМЖ/РЯ. Интересно отметить, что исследователями не было выявлено ассоциаций патогенных вариантов в анализируемых генах с риском развития РМЖ без семейной истории РЯ, и напротив была обнаружена сильная ассоциативная связь при рассмотрении семей только с РЯ [52]. Данные результаты свидетельствуют в пользу того, что гены RAD51C и RAD51D являются генами восприимчивости к РЯ, но не к РМЖ.
Спектр мутаций в генах RAD51C и RAD51D хорошо охарактеризован в работе M.Suszynska. В совокупности в 23 802 образцах РЯ было идентифицировано 46 различных мутаций в гене RAD51C. Мутации в данном гене были довольно равномерно распределены по кодирующей последовательности, однако их концентрация была немного выше в центральной части гена. По функциональной роли примерно две трети составляли мутации сдвига рамки считывания и нонсенс мутации, а 27% представляли собой мутации сайта сплайсинга. Четырнадцать мутаций были обнаружены в трех или более случаях. Из них c.706-2A> G/ RAD51C, c.577C> T/ RAD51C, c.224dupA/ RAD51C и c.955C> T/ RAD51C являются наиболее частыми и зарегистрированы в 11, 9, 7 и 6 случаях, соответственно. Все перечисленные мутации по всей видимости характерны для европейской популяции. Ассоциативный анализ подтвердил, что все четыре мутации, включая наиболее частую мутацию сплайсинга c.706-2A> G/ RAD51C, связаны с высоким риском развития РЯ [139].
В гене RAD51С известно несколько мутаций c эффектом основателя, ассоциированных с РЯ. Две повторяющиеся мутации c.93delG и c.837+1G>A были идентифицированы в финской популяции у пациентов, страдающих РМЖ и/или РЯ. Анализ гаплотипов подтвердил их общее происхождение. Оба варианта показали сильную связь с РЯ, а также с РМЖ в контексте семейного анамнеза РЯ [137]. Еще одна мутация сайта сплайсинга c.571+4A>G с эффектом основателя была обнаружена у пациенток с РМЖ и РЯ из Ньюфаундлена (Канада) [140]. Сообщения о патогенном варианте c.774delT/RAD51C с эффектом основателя также были отмечены в шведской популяции [141].
В гене RAD51D было идентифицировано 39 различных мутаций среди 22787 больных РЯ. Около 70% мутаций распределены в области гена, соответствующей АТФ-связывающему домену белка RAD51D. Большинство идентифицированных мутаций представляли собой мутации сдвига рамки считывания (42%), либо нонсенс (42%) мутации, в то время как на долю мутаций сайта сплайсинга приходилось лишь 9%. Четыре патогенных варианта были обнаружены по крайней мере у шести пациентов. Среди них две нонсенс мутации c.694C>T/RAD51D и c.748delC/RAD51D, идентифицированные соответственно в 11 и 6 случаях, а также две мутации сдвига рамки считывания c.270_271dupTA/RAD51D и c.556C>T/RAD51D, выявленные у 7 и 6 неродственных пациенток, соответственно. Три из четырех рекуррентных мутаций были связаны с повышенным риском развития РЯ у лиц европейского происхождения. Тогда как мутация c.270_271dupTA/RAD51D оказалась аллелем высокого риска развития РЯ, специфичной для восточноазиатской популяции [139].Среди обнаруженных мутаций была выявлена одна мутация с эффектом основателя c.576 + 1G> A/ RAD51D, характерная для финской популяции. Ранее эта мутация была обнаружена у 2,9% пациентов с семейным анамнезом РМЖ и РЯ из Финляндии и по результатам исследования случай-контроль охарактеризована как мутация высокого риска РЯ [138].
Таким образом, несмотря на низкую частоту встречаемости герминальных патогенных мутаций в генах RAD51C и RAD51D, эти паралоги вносят существенный вклад в развитие семейных форм РЯ. Кроме того, имеются данные, что RAD51C- и RAD51D-дефицитные опухолевые клетки могут проявлять чувствительность к ингибиторам PARP [142, 143]. В этом случае скрининг мутаций в этих генах может иметь клиническую ценность для больных раком яичников, обеспечивая более индивидуальное клиническое ведение и лечение.
Ген BRIP1
BRIP1, также известный как белок группы J анемии Фанкони (FANCJ) или связанная с BRCA1 С-концевая геликаза (BACH1), был впервые идентифицирован с помощью тандемной масс-спектрометрии по его физическому взаимодействию с белком BRCA1 [144]. В комплексе с BRCA1 BRIP1 участвует в подавлении роста опухолей и репарации двухцепочечных разрывов ДНК (DSB) во время G2-M фазы клеточного цикла [145]. Ген BRIP1 экспрессируется как в нормальных, так и в злокачественных клетках и контролирует целостность генома посредством регуляции процессов репликации и гомологичной рекомбинации [146].
Герминальные мутации в гене BRIP являются наиболее часто обнаруживаемыми изменениями при раке яичников после мутаций в генах BRCA1/2 и встречаются примерно в 0,6-0,9% случаев эпителиального рака яичников [147]. Предполагаемый кумулятивный риск развития рака яичников у носителей мутаций в гене BRIP1 к 80 годам составляет примерно 5,8 % [127].
По данным недавно проведенного метаанализа, основанном на сравнении ~ 29400 пациентов с РЯ без учета семейной истории из 63 исследований и ~ 116000 контрольных пациентов из базы данных gnomAD, частота патогенных мутаций в гене BRIP1 составила 0,89 %, а риск развития РЯ у носителей этих изменений был повышен практически в пять раз [139]. Однако риск может быть значительно выше при наличии родственников, страдающих РМЖ и/или РЯ. Согласно N.Weber-Lassalle, мутации, приводящие к потере функции белка BRIP1, связаны с 20-кратным повышением риска развития РЯ у пациенток с отягощенным семейным анамнезом. Интересно отметить, что частота встречаемости патогенных мутаций в данном гене среди пациенток исключительно с семейной историей РМЖ оказалась близкой к таковой в контрольной группе. Тогда как среди женщин с РМЖ, имевших в анамнезе случаи РЯ, распространенность мутаций в гене BRIP1 была достоверно выше, чем в контроле [148].
Мутационный спектр в гене BRIP1 наиболее полно охарактеризован в работе M.Suszynska на основе метаанализа, объединившего данные из 44 исследований. В совокупности у 122494 пациенток с РЯ было идентифицировано 71 различный патогенный вариант в данном гене. Мутации были равномерно распределены по большей части кодирующей последовательности гена. Наибольшую долю мутаций составили мутации сдвига рамки считывания (52%), нонсенс мутации (30%) и мутации сплайсинга (15%). Было выявлено пятнадцать рекуррентных мутаций, которые были обнаружены у трех и более неродственных пациенток. Наиболее часто в группе больных РЯ встречались следующие изменения: c.2392C> T/ BRIP1 (0,062%), c.2255_2256delAA/ BRIP1 (0,031%), c.394dupA/ BRIP1 (0,026%), c.2010dupT/ BRIP1 (0,026%) и c.2108_2109insCC/ BRIP1 (0,026%). Все перечисленные мутации были выявлены преимущественно в европеоидной популяции. Для 8 рекуррентных мутаций, идентифицированных как в группе больных РЯ, так и в контроле, были рассчитали OR, специфичные для каждой мутации. Так мутации c.394dupA, c.1236delA, c.2010dupT, c.2255_2256delAA, c.1871C >A и c.2108_2109insCC были связаны с высоким риском развития РЯ. Тогда как наиболее частая мутация c.2392C>T/BRIP1 оказалась аллелем среднего риска [139].
На сегодняшний день широко обсуждается перспектива использования таргетной терапии при лечении РЯ, вызванного потерей функции гена BRIP1. Ранее были получены данные, о том, что наличие мутаций, приводящих к нарушениям геликазной активности белка BRIP1, придавало опухолевым клеткам чувствительность к алкилирующим агентам, таким как цисплатин [149]. В то же время дефицит белка BRIP1, по-видимому, не придает клеткам чувствительности к ингибиторам PARP (PARPi) [150]. Однако до внедрения рекомендаций по лечению карциномы яичников с мутациями в гене BRIP1 необходимо проведение дальнейших исследований по оценке эффективности различных препаратов и их комбинаций.
Роль других генов пути АФ в формировании предрасположенности к РЯ до конца не изучена и является предметом обсуждения. В недавнем исследовании 14 генов пути АФ (FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG/XRCC9, FANCI, FANCL/PHF9, FANCM, FANCP/SLX4, FANCQ/ERCC4, FANCR/RAD51 и FANCU/XRCC2) были изучены на предмет наличия патогенных мутаций в выборке больных с наследственными формами рака (n=1021) и в группе контроля. В результате достоверные ассоциации с риском развития наследственного РМЖ и РЯ были выявлены лишь для гена FANCA [151]. В более раннем исследовании, проведенном E.Dicks с соавторами, ученые установили, что патогенные варианты в гене FANCM способствуют развитию серозного РЯ высокой степени злокачественности (p=0,008), но по-видимому не связаны с развитием других гистотипов РЯ [152]. Подобное исследование было проведено H. Song с соавторами. Ученые провели поиск ассоциаций патогенных вариантов в 54 генах-кандидатах РЯ, в том числе 9 генов пути АФ (PALB2, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCG, FANCI и FANCL), с развитием РЯ высокой степени злокачественности, однако статистически-достоверная связь была показана лишь для гена PALB2 [153].
Заключение. Злокачественные новообразования яичников составляют около 25% от всех злокачественных опухолей женских половых органов, при этом являются главной причиной смертности онкогинекологических больных во многих странах мира, включая Россию. Важнейшая роль в формировании данной онкопатологии отводится генетическим факторам. Наследственные формы РЯ составляют более одной пятой (около 23%) случаев злокачественных новообразований яичников. В настоящее время идентифицировано, по крайней мере, шесть наследственных синдромов, вызванных повреждениями в различных генах и проявляющихся семейной предрасположенностью к возникновению рака органов женской репродуктивной системы. Известно, что около 65-85% наследственных опухолей яичников обусловлены герминальными мутациями в генах BRCA1/2, которые вызывают дефекты репарации ДНК. Однако на сегодняшний день учеными из разных стран выявлено по меньшей мере 16 генов, включая BRCA1, BRCA2, ATM, BARD1, BRIP1, CHEK2, MLH1, MRE11A, MSH2, MSH6, NBN, PALB2, PTEN, RAD51D, RAD51C, STK11, участвующих в механизме канцерогенеза яичников. Подводя итог можно заключить, что в последние годы знания о молекулярных механизмах опухолеобразования яичников существенно расширились, однако многие детали этого процесса остаются не до конца ясными. Изучение генетических и этноспецифических особенностей семейных форм заболевания является на сегодняшний день перспективной областью исследований, результаты которых позволят повысить эффективность диагностики и лечения данной группы злокачественных новообразований и приблизить человечество к прецизионной медицине.
Информация о финансировании
Исследование поддержано программой развития биоресурсных коллекций ФАНО. Работа выполнена при финансовой поддержке гранта РФФИ № 20-34-90003; государственного задания Министерства науки и высшего образования РФ №075-03-2021-193/5; гранта РФФИ № 18-29-09129.
Благодарности
Валова Я.В. выражает благодарность фонду РФФИ (проект № 20-34-90003) за финансовую поддержку данной работы. Прокофьева Д.С. выражает благодарность Министерства науки и высшего образования РФ (гос. задание №075-03-2021-193/5) за финансовую поддержку данной работы. Мингажева Э.Т. выражет благодарность фонду РФФИ (проекта № 18-29-09129) за финансовую поддержку данной работы













Список литературы