
Использование метода Multifactor Dimensionality Reduction (MDR) и его модификаций для анализа ген-генных и генно-средовых взаимодействий при генетико-эпидемиологических исследованиях (обзор)
Aннотация
Актуальность: При генетико-эпидемиологическом исследовании мультифакториальных признаков (заболеваний) важной задачей является оценка ген-генных и генно-средовых взаимодействий, ассоциированных с изучаемым фенотипом. Цель исследования:Провести систематический анализ данных, имеющихся в современной литературе, о возможностях метода Multifactor Dimensionality Reduction (MDR) и его различных модификаций (GMDR, MB-MDR) при изучении ген-генных и генно-средовых взаимодействий. Материалы и методы:В обзор включены современные данные зарубежных и отечественных статей, найденные в Pubmed по данной теме. Результаты:Метод MDR дает возможность оценивать ген-генные и генно-средовые взаимодействия, ассоциированные с качественными фенотипами с учетом коррекции на качественные ковариаты и проводить их валидацию с помощью пермутационного теста. Так же он позволяет проводить кросс-валидацию моделей, оценивать характер (synergy, additive, redundancy) и силу (доля вклада в энтропию) этих взаимодействий и их визуализировать графически. Данный метод не дает возможность изучать количественные фенотипы и учитывать количественные ковариаты. Метод MB-MDR позволяет анализировать межгенные и генно-средовые взаимодействия, ассоциированные с качественными и количественными фенотипами, учитывать в анализе ковариаты, проводить валидацию полученных моделей с помощью пермутационного теста, а также определять отдельные комбинации факторов, ассоциированные с исследуемыми фенотипами с учетом ковариат и значимости (рисковое или протективное значение). Метод GMDR дает возможность оценивать ген-генные и генно-средовые взаимодействия, ассоциированные с качественными фенотипами с учетом коррекции на качественные и количественные ковариаты, проводить их валидацию с помощью пермутационного теста и визуализировать графически, позволяет проводить кросс-валидацию наиболее значимых моделей с учетом коррекции на ковариаты и множественные сравнения (пермутационный тест). Заключение:При генетико-эпидемиологическом исследовании наиболее оптимальным является использование вначале метода MB-MDR для установления наиболее значимых SNP×SNP и генно-средовых взаимодействий, их валидация с помощью пермутационного теста, а также определение конкретных комбинаций, ассоциированных с исследуемым фенотипом. Далее с помощью метода GMDR проведение кросс-валидации наиболее значимых моделей с учетом коррекции на ковариаты и множественные сравнения (пермутационный тест). Затем использование метода MDR для оценки характера (synergy, additive, redundancy) и силы (доля вклада в энтропию) SNP×SNP и генно-средовых взаимодействий и их графической визуализации.
Ключевые слова: полиморфизм, ассоциации, SNP×SNP взаимодействия, генно-средовые взаимодействия, MDR, MB-MDR, GMDR
При генетико-эпидемиологическом исследовании мультифакториальных признаков важной задачей является оценка ген-генных и генно-средовых взаимодействий, ассоциированных с развитием исследуемого фенотипа. Это связано с тем, что наряду с главными эффектами полиморфных локусов значимую роль при формировании мультифакториальных фенотипов (как качественных – например, развитие заболевания, так и количественных, например уровень артериального давления или возраст менархе) имеют ген-генные (эпистатические) взаимодействия и генно-средовые взаимодействия (взаимодействия полиморфных локусов со средовыми факторами риска), вклад которых в развитие сложно наследуемых признаков весьма существенен [1-3]. Существуют различные методы оценки ген-генных и генно-средовых взаимодействий, вовлеченных в развитие как качественных, так и количественных признаков (регрессионный анализ, метод монте-карло марковскими цепями, снижения размерности) [4-8].
Одним из методов, позволяющих оценить межгенные взаимодействия, является регрессионная модель анализа взаимодействия генов, реализованная в программном обеспечении gPLINK v2.050 в рамках использования процедуры «epistasis» [4]. Для оценки бивариантного признака (например, «больной-здоровый») используется модель логистической регрессии, предполагающая, что вероятность события (формирование заболевания) описывается как логическая функция от линейной комбинации независимых переменных (факторов-предикторов, в данном случае полиморфных локусов). При анализе количественных фенотипов (уровень артериального давления, возраст менархе, рост, вес тела, индекс массы тела и др.) используется линейная регрессия от факторов-предикторов. При необходимости в регрессионный анализ включаются ковариаты. Следует отметить, что ограничением данного метода является возможность анализа только двух-локусных взаимодействий и отсутствие процедуры валидизации результатов с учетом множественных сравнений. Изучение эпистатических взаимодействий полиморфных локусов с использованием программного обеспечения gPLINK v2.050 посвящены многочисленные работы [9, 10].
Другим методом, используемым для решения задачи оценки ген-генных взаимодействий, является метод Монте-Карло Марковскими цепями (Markov Chain Monte Carlo, MCMC), реализованный в программном обеспечении APSampler (http://sources.redhat.com/cygwin). В рамках данной программы осуществляется поиск генетических паттернов (сочетания генетических вариантов различных локусов), ассоциированных с фенотипическим признаком (заболеванием) [5, 6]. Поиск паттернов осуществляется с помощью процедуры Монте-Карло Марковскими цепями, при этом на каждом шаге рассматривается сразу несколько паттернов, а их набор оптимизируется от шага к шагу с точки зрения вероятности того, что все паттерны из набора независимо друг от друга и одновременно ассоциированы с признаком. Вероятность ассоциации каждого паттерна оценивается непараметрическим критерием Вилкоксона, при этом сравниваемые подгруппы устроены так, что они отличаются носительством только одного паттерна из набора. В результате этого этапа формируется список паттернов, ассоциированных с развитием заболевания. На следующем этапе происходит валидация этих данных. Для каждого паттерна из списка вычисляется значимость ассоциации по Фишеру. Далее программа несколько раз перемешивает метку фенотипического признака и еще раз запускает поиск ассоциированных паттернов. Достоверности ассоциации по результатам запусков с перемешанным фенотипом дают распределение достоверностей находок при условии нулевой гипотезы, утверждающей отсутствие действительных ассоциаций в начальных данных. Это нулевое распределение используется для валидации сочетаний, найденных на первом этапе [6]. Сила ассоциации оценивается показателем отношения шансов (OR) с 95% доверительным интервалом (95%CI). Коррекция на множественные сравнения проводится с помощью поправки Бонферрони, метода FDR, пермутационного теста. Следует отметить, что ограничением этого метода является невозможность проведения кофакторного анализа, а также он не позволяет изучать генно-средовые взаимодействия. Программное обеспечение APSampler и используемый в нем метод Монте-Карло Марковскими цепями (Markov Chain Monte Carlo, MCMC) для решения задач оценки ген-генных взаимодействий активно применяется в исследовательских работах [11-14].
Широкое распространение в генетико-эпидемиологических исследованиях как зарубежных [7, 15-28] так и российских ученых [29, 30] в последние годы получает метод снижения размерности – MDR (Multifactor Dimensionality Reduction) и его модификации (GMDR, MB-MDR). Данный метод, дает возможность оценить как ген-генные, так и генно-средовые взаимодействия, ассоциированные с формированием мультифакториального фенотипа. Метод снижения размерности MDR (Multifactor Dimensionality Reduction) позволяет уменьшить размерность числа рассчитываемых параметров при одновременной оценке взаимодействий большого количества полиморфизмов за счет конструирования новых переменных на основе суммирования сочетаний генотипов повышенного и пониженного риска формирования заболевания [7, 8, 31].
Метод MDR является непараметрическим [8], он не предполагает какой-то модели наследования признака и поэтому может быть использован для анализа любых фенотипов без априорных предположений. Алгоритм проведения MDR метода представлен следующими этапами. На первом этапе MDR-анализа исходные данные случайным образом делятся на две выборки: обучающую (9/10 данных) и тестовую (1/10 данных). Затем, для каждой комбинации аллелей и генотипов, присутствующей в обучающей выборке, рассчитывается параметр, характеризующий соотношение количества больных и здоровых, несущих эту комбинацию, и в зависимости от величины этого параметра комбинации классифицируются на категории высокого и низкого риска. В результате этого осуществляется переход от n-мерного пространства всех единичных полиморфных участков и фенотипа к двумерному пространству, где одно измерение – это уровень риска, а второе – носительство данной комбинации аллелей. Среди всех возможных выделяются комбинации с наименьшей ошибкой классификации в обучающей и тестовой выборках. Следует отметить, что деление на группы осуществляется 10 раз на основе изменяемых каждый раз параметров генератора случайных чисел (рисунок 2). При этом показатель согласованности модели (Cross Validation Consistency – CVC) показывает, сколько раз из этих 10 она идентифицировалась как лучшая. В результате MDR анализа выявляются наилучшие модели для 2n, 3n, 4n и т.д. сочетаний SNP т.е. модели, имеющие наибольший показатель согласованности (CVC) и наименьшую ошибку предсказания (наибольшую точность предсказания модели – Testing Balanced Accuracy). Модель считается валидированной, если ее согласованность 9/10 и более [7, 8, 31].
Метод MDR реализован в программном обеспечении MDR (http://sourceforge.net/projects/mdr). Коррекция на множественные сравнения в данном случае проводится с помощью пермутационного теста в программном обеспечении MDRpt (http://sourceforge.net/projects/mdr/files/mdrpt). Следует отметить, что программное обеспечение метода MDR позволяет построить дендрограмму или граф и таким образом визуализировать рассматриваемые ген-генные и генно-средовые взаимодействия, а также оценить характер этих взаимодействий (synergy, additive, redundancy) и их силу (доля вклада в энтропию). В работе Москаленко М.И. [32] представлены данные, полученные с использованием метода MDR при анализе взаимодействия полиморфных локусов матриксных металлопротеиназ (MMP), ассоциированных с развитием гипертонической болезни. На рисунке 1 представлены данные, характеризующие комбинации генотипов повышенного (темно-серые ячейки), пониженного (светло-серые ячейки) и отсутствия (белые ячейки) риска в рамках наиболее значимых моделей межгенных взаимодействий (А, В, С, D – обозначение моделей). На рисунке 2 представлены дендрограмма (А) и граф (В) межгенных взаимодействий ММР при формировании эссенциальной гипертензии, демонстрирующие характер этих взаимодействий (synergy, additive, redundancy) и их силу (доля вклада в энтропию).
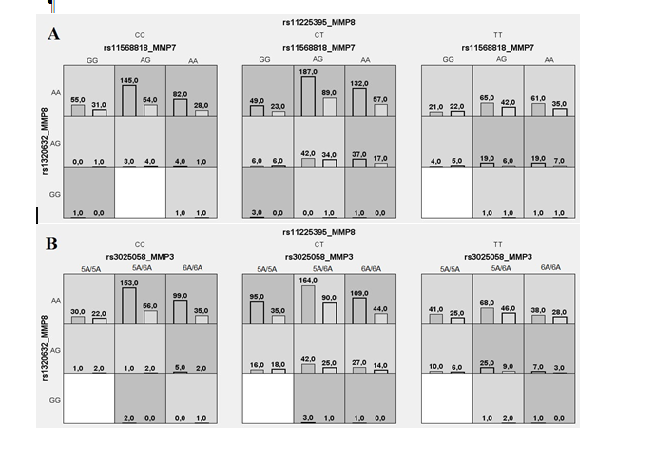




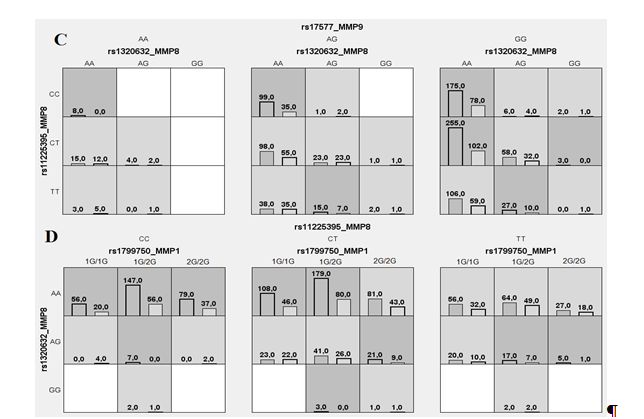
Рис. 1. Диаграмма моделей взаимодействий полиморфных локусов ММР при формировании эссенциальной гипертензии (ЭГ)
Примечание: получено методом MDR, здесь и далее темно-серые ячейки – комбинации повышенного риска, светло-серые – пониженного риска, белые – сочетания комбинаций генотипов отсутствуют. Столбики слева – количество больных с ЭГ, столбики справа – число индивидуумов контрольной группы; А, В, С, D – обозначение моделей межгенных взаимодействий (данные получены Москаленко М.И [32]).
Fig. 1. Diagram of interaction models of polymorphic MMP loci during the formation of essential hypertension (EH)
Note: obtained by the MDR method, hereinafter, dark gray cells – combinations of increased risk, light gray – reduced risk, white – combinations of combinations of genotypes are absent. The bars on the left are the number of patients with EH, the bars on the right are the number of individuals in the control group; A, B, C, D – designation of models of intergenic interactions (data obtained by Moskalenko M.I. [32]).


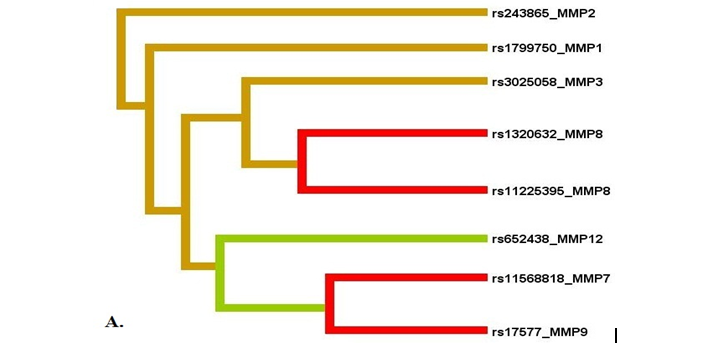
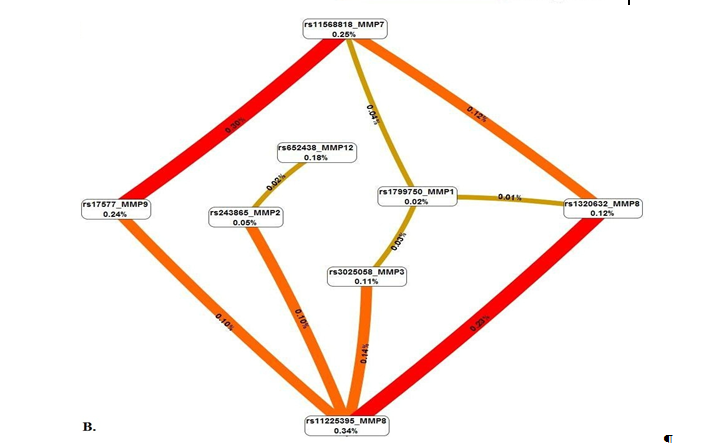
Рис. 2. Дендрограмма (А) и граф (В) межгенных взаимодействий ММР при формировании эссенциальной гипертензии
Примечание: характер взаимодействия между ММР при формировании фенотипа характеризуется цветом линии: красный – выраженный синергизм, оранжевый – умеренный синергизм, синий – коричневый – аддитивное взаимодействие. Сила и направленность взаимодействия выражены в % энтропии (данные получены Москаленко М.И [32]).
Fig. 2. Dendrogram (A) and graph (B) of intergenic interactions of MMPs during the development of essential hypertension
Note: the nature of the interaction between the MMP in the formation of the phenotype is characterized by the color of the line: red – pronounced synergism, orange – moderate synergism, blue – brown – additive interaction. The strength and direction of interaction are expressed in% entropy (data obtained by Moskalenko M.I. [32]).
Таким образом, метод MDR дает возможность оценивать ген-генные и генно-средовые взаимодействия, ассоциированные с качественными фенотипами с учетом коррекции на качественные ковариаты и проводить валидацию с помощью пермутационного теста. Так же он позволяет проводить кросс-валидацию моделей (определение показателей согласованности (CVC), точности предсказания (Testing Balanced Accuracy), чувствительности (Se) и специфичности (Sp) моделей), оценивать характер (synergy, additive, redundancy) и силу (доля вклада в энтропию) SNP×SNP и генно-средовых взаимодействий и их визуализировать в виде графа. При этом, данный метод не дает возможность изучать количественные фенотипы и учитывать количественные ковариаты, а также получать точное значение рperm.
К настоящему времени разработано достаточно большое количество модификаций метода MDR – Generalized MDR (GMDR), Pedigree-based GMDR (PGMDR), Cox-based MDR (Cox-MDR), Pair-wise MDR (PW-MDR), Quantitative MDR (QMDR) и др. [7], адаптированные под отдельные программные оболочки (MATLAB, Java, Python, R и др.) и характеризующиеся своими «исследовательскими» особенностями: возможность учитывать качественные и количественные ковариаты при анализе, изучение небольших обьемов выборок, исследование качественных и количественных фенотипов, проведение пермутационных процедур и т.д. С более детальными характеристиками этих модификаций метода MDR можно ознакомиться в работе Gola D et al. [7]. В настоящей работе мы рассмотрим две модификации метода MDR – MB-MDR и GMDR, которые мы использовали в наших исследованиях.
Метод Model-Based-MDR (MB-MDR), в отличие от MDR, позволяет анализировать межгенные взаимодействия, ассоциированные как с качественными (развитие заболевания), так и с количественными признаками, учитывать в анализе ковариаты и проводить валидацию полученных моделей с помощью пермутационного теста. Следует отметить, что метод MB-MDR, отличается высокой статистической мощностью при наличии генетической гетерогенности [33, 34]. При проведении MD-MDR анализа выделяются 3 категории генотипов – высокого риска, низкого риска и не влияющие на риск. Далее каждая группа комбинаций генотипов сравнивается с двумя другими группами и результатом этого является выделение нескольких комбинаций генотипов, связанных (согласно статистики Вальда и уровня значимости ассоциации) с исследуемым фенотипом. Далее для каждой тестируемой комбинации выбирается вариант, соответствующий максимальному значению статистики Вальда и затем определяется экспериментальный уровень значимости с помощью пермутационного теста.
При использовании метода MD-MDR наиболее оптимальной является кодоминантная схема кодирования полиморфных локусов, обеспечивающая наилучший баланс между уровнем ошибки 1-го рода и мощностью исследования [35, 36]. В рамках метода MD-MDR проводится тестирование как правило двух-, трех- и четырехлокусных (факторных) комбинаций. Хотя данный метод позволяет учитывать комбинации и большего числа факторов. При необходимости в анализ включаются ковариаты (как качественные, так и количественные). В конечном итоге рассматриваются модели (в среднем 3-4 модели каждого уровня) с наибольшими статистиками Вальда и уровнем значимости. Валидация результатов проводится с помощью пермутационного теста. Также, с помощью метода MB-MDR устанавливаются отдельные комбинации генотипов (и факторов риска), ассоциированные с исследуемыми фенотипами, с расчетом их статистической значимости. Метод MB-MDR (Model Based Multifactor Dimensionality Reduction) реализован в программном обеспечении MB-MDR (Version 2.6) для программной среды R.
Таким образом, метод MB-MDR позволяет анализировать межгенные и генно-средовые взаимодействия, ассоциированные с качественными и количественными фенотипами, учитывать в анализе ковариаты, проводить валидацию полученных моделей с помощью пермутационного теста, а также определять отдельные комбинации генотипов (и факторов среды), ассоциированные с исследуемыми фенотипами с учетом их уровня значимости. При этом, программное обеспечение метода MB-MDR не дает возможность визуализировать графически (в виде графа или дендрограммы) установленные ген-генные и генно-средовые взаимодействия.
Метод Generalized MDR (GMDR) [37, 38] (http://www.ssg.uab.edu/gmdr), реализованный в программном обеспечении GMDR (software Beta 0.9) (http://sourceforge.net/projects/gmdr) является модификацией метода MDR и основан на тех же принципах что и этот метод. Он позволяет оценивать ассоциации качественных фенотипов с различными комбинациями генотипов и факторов среды в рамках 2-х, 3-х, n-локусных моделей. Следует отметить, что программное обеспечение метода GMDR позволяет построить граф (дендрограмму) и таким образом визуализировать рассматриваемые ген-генные и генно-средовые взаимодействия, а также оценить характер этих взаимодействий (synergy, additive, redundancy). При расчетах метод GMDR позволяет учитывать как качественные, так и количественные ковариаты. Данный метод дает возможность проводить кросс-валидацию наиболее значимых моделей с расчетом показателей согласованности (CVC), точности предсказания (Testing Balanced Accuracy), чувствительности (Se) и специфичности (Sp) моделей, при этом так же учитываются ковариаты. Коррекция на множественные сравнения проводится с помощью пермутационного теста в Perl script (“perl GMDR_permutatin.pl”) программного обеспечения GMDR (software Beta 0.9). При этом указывается необходимое количество пермутаций (разработчики метода GMDR отмечают, что для получения р-значения на уровне 0,001 необходимо 1000 пермутаций, а для получения р-значения на уровне 0,0001 должно быть 10000 пермутаций) и необходимой число кросс-валидаций (разработчики указывают, что при обьеме выборки до 500 человек достаточно 5 кросс-валидаций, а при выборке более 1000 человек необходимо 10 кросс-валидаций). При пермутационном тесте так же возможна коррекция на ковариаты. Следует отметить, что метод GMDR не позволяет изучать ген-генные и генно-средовые взаимодействия, ассоциированные с количественными признаками.
В таблице 1 и на рисунке 3 представлены данные, полученные Милановой С.Н. [39] с использованием метода GMDR при анализе генно-средовых взаимодействий полиморфных локусов цитокинов с употреблением алкоголя, ассоциированных с развитием инсульта на фоне гипертонической болезни. В таблице приведены наиболее значимые модели генно-средовых взаимодействий, ассоциированные с развитием инсульта на фоне гипертонической болезни, а также результаты их кросс-валидации (показатели воспроизводимости моделей (CVC) и точности предсказания моделей (Test Bal. Acc.)) и пермутационных процедур. На рисунке 6 представлены дендрограмма (А) и граф (В) генно-средовых взаимодействий, ассоциированные с развитием инсульта на фоне гипертонической болезни, демонстрирующие характер этих взаимодействий (synergy, additive, redundancy) и их силу (доля вклада в энтропию).
Таблица 1
Наиболее значимые модели генно-средовых взаимодействий полиморфных локусов цитокинов с употреблением алкоголя, ассоциированные с развитием инсульта на фоне гипертонической болезни
Table 1
The most significant models of gene-environment interactions of polymorphic cytokine loci with alcohol use, associated with the development of stroke associated with hypertension
Модель | Модель генно-средовых взаимодействий | OR (95%CI) | Test Bal. Acc. | р |
А | ALK×rs1800629 TNFa×rs767455 TNFR1× rs1061624 TNFR2×rs909253 Ltα | 3,84 (2,40-6,14) | 59,42 | 0,001 |
B | ALK×rs1800629 TNFa×rs767455 TNFR1× rs1061624 TNFR2×rs6214 IGF1 | 3,25 (2,06-5,12) | 53,29 | 0,011 |
Примечание: получены в программе GMDR с коррекцией на уровни ТГ, ЛПВП, курение, подверженность частым стрессовым ситуациям; воспроизводимость моделей (CVC) составила 100%; Test Bal. Acc. – точность предсказания модели (%), проведен пермутационный тест – выполнено 1000 пермутаций при 10 кросс-валидациях, что обеспечивает рperm<0,001 (данные получены Милановой С.Н. [39]).
Note: obtained in the GMDR program with correction for levels of TG, HDL, smoking, exposure to frequent stressful situations; model reproducibility (CVC) was 100%; Test Bal. Acc. – model prediction accuracy (%), permutation test was carried out – 1000 permutations were performed with 10 cross-validations, which ensures perm <0.001 (data obtained by Milanova S.N. [39]).
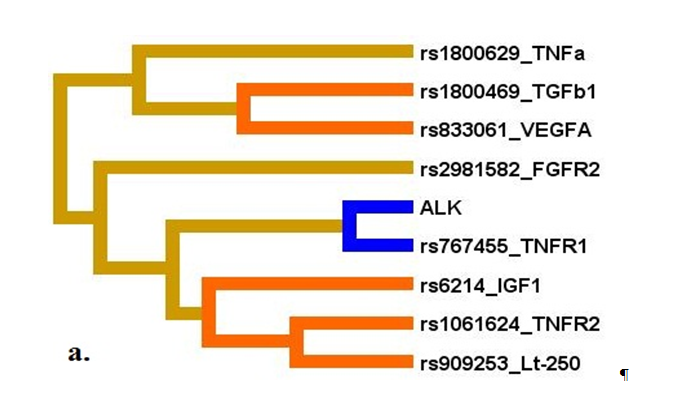


 Рис. 3. Дендрограмма (А) и граф (В) генно-средовых взаимодействий полиморфных локусов цитокинов и употребления алкоголя при формировании ишемического инсульта на фоне гипертонической болезни
Рис. 3. Дендрограмма (А) и граф (В) генно-средовых взаимодействий полиморфных локусов цитокинов и употребления алкоголя при формировании ишемического инсульта на фоне гипертонической болезни
Примечание: направленность взаимодействий между генами-кандидатами при формировании фенотипа обозначаются линиями разного цвета: красного – выраженный синергизм, оранжевого – умеренный синергизм, синего – выраженный антагонизм, зеленого – умеренный антагонизм, коричневого – аддитивное взаимодействие. Сила и направленность взаимодействий представлены в % энтропии (данные получены Милановой С.Н. [39]).
Fig. 3. Dendrogram (A) and graph (B) of gene-environmental interactions of polymorphic loci of cytokines and alcohol use during the development of ischemic stroke associated with hypertension
Note: the orientation of the interactions between candidate genes during the formation of the phenotype is indicated by lines of different colors: red – pronounced synergism, orange – moderate synergism, blue – pronounced antagonism, green – moderate antagonism, brown – additive interaction. The strength and direction of interactions are represented in% entropy (data obtained by Milanovoy S.N. [39]).
Таким образом, метод GMDR дает возможность оценивать ген-генные и генно-средовые взаимодействия, ассоциированные с качественными фенотипами с учетом коррекции на качественные и количественные ковариаты, проводить валидацию с помощью пермутационного теста, оценивать характер (synergy, additive, redundancy) и силу (доля вклада в энтропию) SNP×SNP и генно-средовых взаимодействий и их визуализировать в виде графа. Так же метод GMDR позволяет проводить кросс-валидацию наиболее значимых моделей взаимодействий с учетом коррекции на ковариаты и коррекцию на множественные сравнения с помощью пермутационного теста.
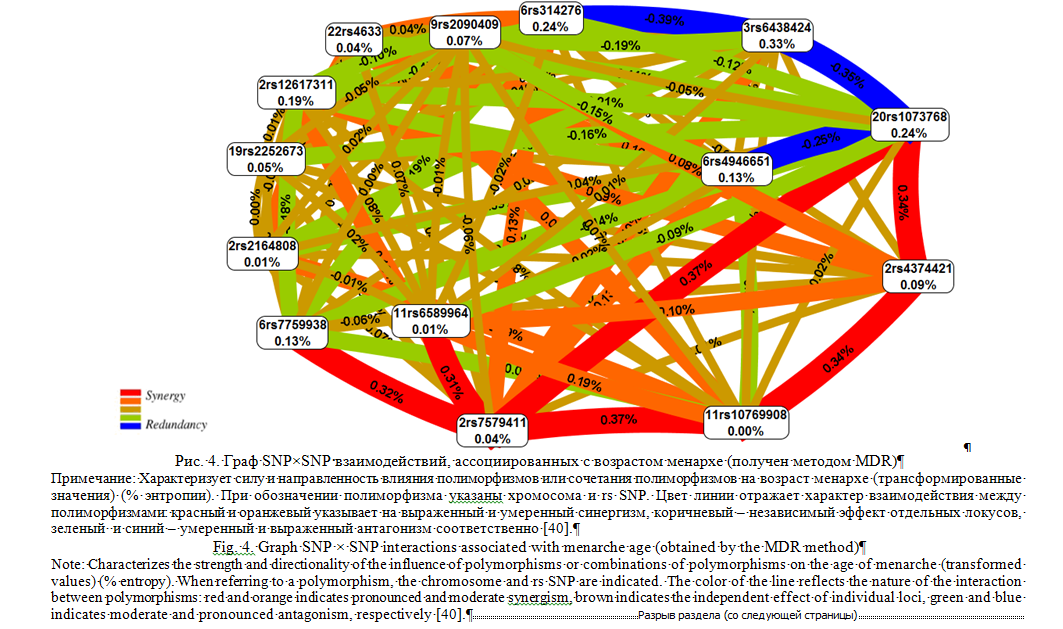
В качестве примера эффективности использования метода MDR и его модификации MB-MDR для оценки ген-генных взаимодействий, ассоциированных с формированием количественных мультифакториальных признаков, приведем полученные нами результаты генетико-эпидемиологического исследования возраста менархе, опубликованные в журналах GENE [40]. Нами с помощью метода MB-MDR изучены ассоциации ген-генных взаимодействий 52 SNPs с возрастом менархе у женщин России. Выборка для исследования составила 1613 женщин. Для генетико-статистического анализа использовались трансформированные значения возраста менархе в связи с тем, что его распределение в исследуемой выборке отличалось от нормального. Изучение межгенных взаимодействий проводилось с коррекцией на коварианты – год рождения (дискретная переменная) и наличие изолированных или сочетанных доброкачественных опухолей и гиперпластических процессов матки (дискретная переменная). В работу включались наиболее значимые модели ген-генных взаимодействий: p<1*10-4 для двухлокусных моделей, p<1*10-7 для трехлокусных моделей, p<1*10-12 для четырехлокусных моделей. Коррекция на множественные сравнения проводилась с помощью пермутационного теста (выполнялось 1000 пермутаций) для отобранных для исследования наиболее значимых 2-х, 3-х и 4-х локусных моделей. Статистически значимыми считали модели с pperm<0,01. Оценка характера (synergy, additive, redundancy) и силы (доля вклада в энтропию) межгенных взаимодействий, а также их визуализация в виде графа проводилась с помощью метода MDR. Установлено, что 14 полиморфных локусов из 52 изученных SNPs ассоциированы с возрастом менархе в составе 12 наиболее значимых 2-х, 3-х и 4-х локусных моделей ген-генных взаимодействий (pperm≤0,006). Выявлены антагонистические взаимодействия 4 полиморфных локусов (определяют -0,25 - -0,39% энтропии признака) (рисунок 4) - rs6438424 3q13.32, rs1073768 GHRH, rs4946651 LIN28B, rs314276 LIN28B (на рисунке линии их объединяющие окрашены в синий цвет) и синергизм (детерминируют 0,31-0,37% энтропии) в ген-генных взаимодействиях 6 SNPs - rs1073768 GHRH, rs7579411 LHCGR, rs7759938 LIN28B, rs4374421 LHCGR, rs6589964 BSX, rs10769908 STK33 (на рисунке линии их объединяющие окрашены в красный цвет).
Итак, проведенный анализ литературных данных позволяет заключить, что при изучении ассоциаций полиморфных локусов с мультифакториальными признаками (как качественными – заболеваниями, так и количественными) наиболее оптимальным является использование вначале метода MB-MDR для установления наиболее значимых SNP×SNP и генно-средовых взаимодействий, их валидация с помощью пермутационного теста, а также определение конкретных комбинаций, ассоциированных с развитием исследуемого фенотипа. Далее с помощью метода GMDR проведение кросс-валидации наиболее значимых моделей взаимодействий с учетом коррекции на ковариаты и коррекцию на множественные сравнения с помощью пермутационного теста. Затем использование метода MDR для оценки характера (synergy, additive, redundancy) и силы (доля вклада в энтропию) SNP×SNP и генно-средовых взаимодействий и их визуализации в виде графа.
В отношении данной статьи не было зарегистрировано конфликта интересов.













Список литературы