
Familial translocation t(5;11)(q32;q23) resulting in Jacobsen syndrome and distal trisomy 5q31 in a female patient
Aннотация
Background: Jacobsen syndrome is a rare contiguous gene disorder caused by partial deletion of the distal part of the long arm of chromosome 11 (prevalence 1/100,000 live births). Most of the reported cases are caused by de novo deletion and in 15% result from an unbalanced segregation of a familial balanced translocation. The spectrum of clinical symptoms observed in patients with JBS is variable and depends on the size of the deletion. The aim of the study: To describe the phenotypic variation in a patient with Jacobsen syndrome as a result of the segregation of an unusual balanced translocation inherited from her mother. Materials and methods: A detailed clinical analysis of the patient’s condition was performed. The chromosomes were studied through the GTG-banding analysis and FISH withCri-du-Chat Region Probe – LSI EGR1, LSI D5S23, D5S721. Results: The patient has a derivative chromosome 11 with a partial monosomy 11q and a partial trisomy 5q with a phenotype influenced by these two imbalances. The mother showed a 46,XX,t(5;11)(q32;q23) karyotype. Conclusion: Deletion syndromes arising from apparently balanced translocation are unusual ways of presentation in Jacobsen syndrome and should be evaluated carefully when the distal part of chromosome 11q is involved in the rearrangement.
Introduction. Jacobsen syndrome (syndrome by deletion11q, OMIM no. 147791) is a very rare but well-known entity caused by partial deletion in the distal region of the long arm of chromosome 11. The incidence is reported to be 1 per 100,000, and females are more affected than males in a rate 2:1. Jacobsen syndrome manifests itself with a variable phenotypic expression including developmental and growth delay before and after birth, thrombocytopenia (type Paris-Trousseau) or pancytopenia, heart defects, trigonocephaly and craniofacial malformations with ocular hypertelorism, short nose, broad nasal bridge, anteverted nares, v-shaped mouth and low set malformed ears [1-3].
A minority of patients were reported with partial monosomy of chromosome 11qare unbalanced products of a parental translocation (15% of cases) [1]. Here we report on a Jacobsen syndrome case in an unusual presentation due to a derivative chromosome carried by the mother causing in the patient a distal trisomy to 5q32.
The aim of the study. Here we aimed to describe the phenotypic variation in a patient with Jacobsen syndrome due to the segregation of an unusual balanced translocation inherited from her mother.
Materials and methods. Cell culture and chromosome preparation was performed according to the standard procedure from blood obtained from the patient and her parents [4] Karyotyping was performed based on GTG-banding.
We used the LSI probe D5S23, D5S721S spectrum Green / LSI EGR1 spectrum Orange probes for Cri-du-Chat syndrome to define the breakpoint in the maternal chromosome 5.
Results and discussion. The pro band was the product of the first pregnancy of non-consanguineous parents, there was no family record of genetic disorders. During pregnancy, the mother had high blood pressure, fetal growth retardation and polyhydramnios. Esophageal atresia with fistula was corrected by surgery three days after she was born. During the recovery period she kept a good suction and gained weight.
Since the first evaluation by a genetic specialist, the clinical analysis showed small anterior fontanelle, cephalic circumference of 33 cm, palpebral fissure slant upward, convex nose, long philtrum, thin lips, micrognatia, rotated ears, 4th and 5th metacarpal shortening and long fingers and toes.
At the age of three months, the patient was revaluated and presented a light spasticity, her neurodevelopmental state was normal for her age: she smiled, gurgled and she was starting to support the head. An abdominal ultrasound was performed searching internal malformation, and the result was normal. At one year and a month of life, she was consulted by Genetics services again, and there were observed trigonocephaly, closed anterior fontanels, microcephaly (cephalic circle in 41cm), hypertelorism, high nasal bridge, long philtrum, carp shaped mouth, thin upper lip, slight micrognatia, low-set rotated ears, atypicalcreases of the palms, teletelia and hypotonia.
The 5q31 signal, LSI EGR1 spectrum Orange probes for Cri-du-Chat syndrome, was present at the terminal end of the long arm of the maternal derivative chromosome 5. It means that the breakpoint was distal to this point probably in 5q32.
The patient´s karyotype was 46,XX,der(11)(q32;q23). The mother´s karyotype was 46,XX,t(5;11)(q32;q23) with an apparently balanced translocation between chromosomes 5 and 11 (Fig. 1). The father’s karyotype was normal.
The patient has shown advances in her neurological development although a remarkable language delay and motor skills are still present.
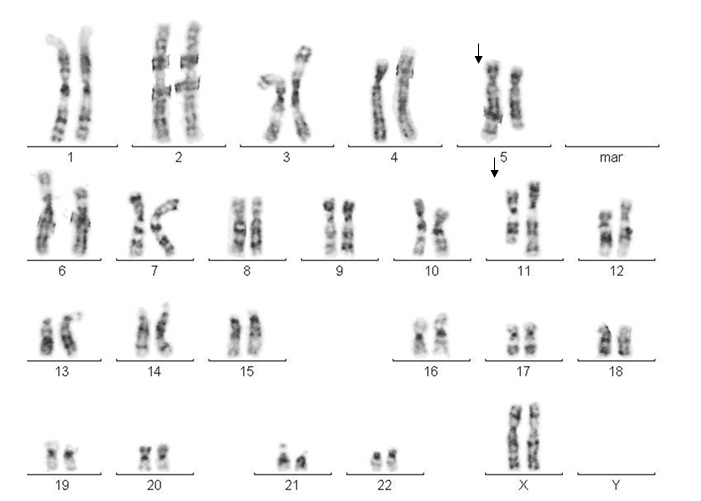
Fig. 1. The mother´s karyotype shows both derivative chromosomes: der(5) y der(11)
We report on a girl in which many of the phenotypic characteristics are coincident for the partial monosomy of 11 and the partial trisomy of 5 (Table 1). However, there are features pertaining only to the partial trisomy of 5. A duplication in this region is a very rare event with almost 40 cases reported in the literature, most of these involving the distal third of 5q (5q31-qter) [5]. Esophageal atresia seems to justify the polyhydramnios present in the prenatal period, which haven´t been reported before in Jacobsen syndrome, although it has been reported for partial trisomy of the region 5q31-35 [6]. Several clinical signs have been described for partial trisomy 5q. Schinzel and colleagues reported four female patients with duplication in the region q31 to q35 of chromosome 5 which dimorphic features are similar to those found in the patient with duplication of segment of smaller size: short stature and Microcephaly, hypertelorism, strabismus, small jaw, low-set ears, highly presence of cardiac defects. In some patients there were reported independence of ptosis of superior eyelid, hypoplasia of the tongue, umbilical hernia, hydronephrosis, postaxial hexadactily and triphalangic thumb
Table 1
Referral reason compared to reported in medical literature for 11q deletion and 5q partial trisomy

Note: * High nasal bridge (lately) and micrognatia are associated with Jacobsen syndrome [1-3]; ** [5, 7]
Our patient has many clinical features pertaining to Jacobsen syndrome (Table 1). However, typical features of this syndromeб such as heart anomalies and / or thrombocytopeniaб are not present in the girl [1, 2]. The breakpoint most frequently involved in the 11q deletion (in 70-80% of cases) is located in the 11q23.3 region [8]. The 11q23-qter region includes 342 genes, and 174 are located in 11q24.1-qter, the critical region established for the expression of Jacobsen syndrome [1].
The finding that our patient does not present thrombocytopenia could be due to the fact that the deletion in 11q does not include the critical region of 6.8 Mb in the terminal portion of 11q (distal to D11S1351), where 4 genes involved in hematopoiesis and platelet function are located (ETS-1, FLI-1, NFRKB and JAM3) [9, 10]. The FLI1 gene, which is the most relevant gene for thrombocytopenia in Jacobsen syndrome, does not appear to be deleted in this patient [11].
Another distinctive feature absent in this patient is the presence of heart disease (which is related to the region between D11S707 and telomere, where more than one gene appears to be involved) [3, 12]. These two characteristics stated above suggest that the 11q deletion of our patient is interstitial and of small size.
Due to the wide variation in the location of the breakpoint in Jacobsen syndrome and the large number of genes showing haploinsufficiency in the critical region, it can be argued that 11q is not a single, clearly defined disease, but a combination of genetic disorders with overlapping phenotypes. There are probably more than 100 genes in the 13.5 MB interval, a significant part of which may exhibit haploinsufficiency. The clinical heterogeneity of this syndrome is quite variable and some reports even described patients with ordinary intelligence [3].
Despite the limitations of this study in that the size of the 11q deletion could not be determined by molecular methods, we consider that the clinical delineation of the patient and the fact of reporting this unusual translocation as a cause of Jacobsen syndrome contributes to showing a different way of presenting this disease.
Conclusion. Deletion syndromes arising from apparently balanced translocation are unusual ways of presentation in Jacobsen syndrome and should be evaluated carefully when the distal part of chromosome 11q is involved in the rearrangement.
No conflict of interest was recorded with respect to this article.
Благодарности
The authors want to recognize the support of the RFBR and CITMA institutions of Russia and Cuba respectively for our study













Список литературы