
Цитогенетический анализ в эпоху высокоразрешающих молекулярно-цитогенетических методов: возможности «обратного» кариотипирования
Aннотация
Актуальность: Внедрение высокоразрешающих молекулярно-цитогенетических методов в клиническую практику позволило определять сложные «скрытые» структурные хромосомные перестройки, не выявленные классическим цитогенетическим анализом. Некоторые из них, размером от 5 млн пар нуклеотидов, можно определить с помощью повторного или «обратного» кариотипирования, проведенного после молекулярных исследований. В этом случае применяется «таргетный» подход к исследованию перестроенной хромосомы на метафазных пластинках с разрешением 500-800 полос на гаплоидный кариотип. «Обратное» кариотипирование необходимо для дальнейшего цитогенетического обследования семьи больного ребенка на носительство возможной сбалансированной хромосомной перестройки, поскольку она не может быть выявлена молекулярным методом. Цель исследования:Выявить методом «обратного» кариотипирования сложные структурные хромосомные перестройки, обнаруженные молекулярно-цитогенетическими методами, у больных детей, первичное кариотипирование которых не определило хромосомные аномалии; провести цитогенетическое и FISH исследования родителям для прогноза будущего потомства. Материалы и методы:Проведено повторное цитогенетическое исследование («обратное» кариотипирование) девяти детям с задержкой психоречевого и психомоторного развития, пороками и/или микроаномалиями развития, имеющим несбалансированные структурные хромосомные (геномные) аномалии, выявленные методом молекулярного кариотипирования. Проведено цитогенетическое и FISH исследования их родителям. В работе были использованы классические цитогенетические методы, FISH исследование, молекулярное кариотипирование с оригинальным биоинформатическим анализом. Результаты:Приведены цитогенетические, молекулярно-цитогенетические и клинические данные о 9-ти пациентах с задержками развития, пороками и/или микроаномалиями развития, имеющих несбалансированные структурные хромосомные аномалии размером от 4,7 млн пар нуклеотидов и более, а также данные об обследовании их родителей. Все девять случаев «скрытых» хромосомных перестроек были выявлены повторным «обратным» кариотипированием. В большинстве случаев аномалия представляла собой изменение дифференциальной исчерченности участка перестройки при неизменённой длине хромосомы. Обследование родителей позволяет проводить корректное медико-генетическое консультирование семьи. Заключение:Применение «обратного» кариотипирования показало его эффективность для детекции небольших по размеру, но цитогенетически видимых, перестроек. Молекулярно-цитогенетические и цитогенетические методы исследования должны использоваться совместно для достижения наиболее корректных результатов генетической диагностики в семье, включая больного ребёнка.
Ключевые слова: «обратное» кариотипирование, молекулярное кариотипирование, «скрытые» хромосомные перестройки, задержка психоречевого (ЗПРР) и психомоторного развития (ЗПМР), микроаномалии развития (МАР)
Введение. Внедрение высокоразрешающих молекулярно-цитогенетических методов в клиническую практику позволило определять сложные «скрытые» структурные хромосомные перестройки, не выявленные классическим цитогенетическим анализом. Некоторые из них, размером от 5 млн пар нуклеотидов (пн), можно выявить с помощью повторного или «обратного» кариотипирования, проведенного после молекулярных исследований. «Обратное» кариотипирование необходимо для дальнейшего цитогенетического обследования семьи больного ребенка на носительство возможной сбалансированной хромосомной аномалии.
При генетическом обследовании детей с недифференцированной задержкой психоречевого, психомоторного развития, пороками и микроаномалиями развития, как правило, первым лабораторным исследованием является цитогенетический анализ. Известно, что максимальная разрешающая способность цитогенетического метода составляет 5-7 млн пн при условии проведения исследования на хромосомных препаратах с разрешением 500-800 полос на гаплоидный кариотип и дифференциального окрашивания хромосом по длине. В некоторых случаях структурные хромосомные аномалии сложны для выявления ввиду малого размера утраченного/дополнительного хромосомного материала или в случаях несбалансированных транслокаций, если фрагменты перестроенных хромосом имеют одинаковый размер [1, 2, 3]. Методы молекулярного кариотипирования (серийная сравнительная геномная гибридизация – arrayCGH и SNParray), вошедшие в арсенал генетических методов исследования, позволяют выявлять геномный дисбаланс от 1000 пн, и с помощью биоинформатического анализа определять гены, вовлеченные в перестройку [4, 5, 6]. Наряду с субмикроскопическими изменениями генома этими методами выявляются и более крупные участки дисбаланса (от 5 млн пн), которые можно обнаружить цитогенетическим анализом. Однако, довольно часто «первичное» кариотипирование, проведенное пациентам в различных лабораториях мира, сложных аномалий хромосом не выявляет. В таких случаях целесообразно проведение повторного («обратного») кариотипирования после проведения молекулярно-цитогенетических методов для «таргетного» исследования аномальной хромосомы. Проведение «обратного» кариотипирования вызвано необходимостью обследования не только больного ребенка, но и его родителей, а также других членов семьи для выявления возможного носительства сбалансированных перестроек, которые нельзя обнаружить молекулярными методами. Для проведения классического кариотипирования членам семьи необходимо знать о хромосомной перестройке у больного ребенка, чтобы провести поиск изменений в кариотипе у родителей. Наиболее эффективными для обнаружения сбалансированных перестроек могут быть цитогенетический и FISH методы [7]. Однако FISH исследование и необходимые ДНК пробы не всегда доступны в отечественных лабораториях, тогда как «таргетное» кариотипирование, проведённое на хромосомных препаратах с разрешением 550 полос и выше при дифференциальном окрашивании, требует минимальных технических и финансовых затрат. Несомненно, в отдельных, особенно сложных случаях, необходимо применение и FISH метода.
С момента внедрения метода молекулярного кариотипирования в лабораторную практику нами было проведено «обратное» кариотипирование детям, у которых размер участков хромосомного нарушения, выявленного при молекулярном кариотипировании, позволял обнаружить хромосомную перестройку при повторном цитогенетическом исследовании.
В данной статье мы представляем 9 случаев повторного «таргетного» цитогенетического анализа после проведения молекулярно-цитогенетического (молекулярное кариотипирование и в некоторых случаях FISH) обследования детей с хромосомными «скрытыми» микроаномалиями, не выявленными при первичном классическом кариотипировании, и обследования их семей.
Цель исследования. Выявить «обратным» кариотипированием сложные структурные хромосомные перестройки, обнаруженные молекулярным кариотипированием, у больных детей, первичный кариотип которых определён без хромосомных аномалий; провести цитогенетическое и FISH исследования родителям для прогноза будущего потомства.
Материалы и методы исследования. Проведены молекулярно-цитогенетические и цитогенетические исследования 9-ти пациентам (детям от 1,5 лет до 14 лет) c ЗПМР, ЗПРР и/или МАР и членам их семей. Молекулярное кариотипирование проводили согласно ранее описанному протоколу, при использовании SNP/олигонуклеотидной микроматрицы c разрешением не менее 1 тысячи пн (Affymetrix). Оценка результатов проводилась с помощью ранее разработанной биоинформатической технологии [5, 6]. Цитогенетический анализ проводился на препаратах метафазных и прометафазных хромосом с разрешением 500-800 полос на гаплоидный кариотип при использовании дифференциального окрашивания хромосом по длине (GTG- и CBG-окрашивание), полученных путем культивирования in vitro лимфоцитов периферической крови в соответствии со стандартной методикой [8, 9]. Культивирование клеток проводилось на питательных средах PB-MAX или RPMI-1640(T), которые позволяют получать хромосомные препараты более высокого разрешения. Колхицин вводили в культуру клеток за 10-20 минут до начала фиксации в конечной концентрации 0,5 мкг/мл. Анализ проводился под световым микроскопом при увеличении х1150 с использованием компьютерной программы анализа изображения. FISH исследования осуществлялись с использованием специфических ДНК проб из коллекции лаборатории молекулярной генетики и цитогеномики мозга им. проф. Ю.Б.Юрова ФГБНУ НЦПЗ по ранее описанным протоколам [10, 11]. Результаты кариотипа представлены согласно международной номенклатуре цитогенетики человека (ISCN) [12].
Результаты и их обсуждение. Во всех девяти случаях при первичном классическом цитогенетическом исследовании, проведенном в различных отечественных и зарубежных лабораториях, хромосомных аномалий у детей выявлено не было, кариотип был записан как 46,ХХ или 46,XY. Методом молекулярного кариотипирования у больных детей были обнаружены структурные несбалансированные перестройки протяженностью от 4,7 млн пн и выше. При повторном цитогенетическом исследовании («обратном» кариотипировании) эти перестройки были визуализированы под микроскопом при увеличении х1150. Также были обследованы родители пробандов для уточнения прогноза потомству. Ниже приводятся результаты исследования 9-ти случаев.
Случай 1.
У мальчика в возрасте 1 год 7 месяцев клинические признаки были следующие: ЗПМР – голову держит с 4 мес., не ползает, не сидит; ЗПРР –речь отсутствует, в том числе нет понимания обращенной речи, не различает родственников, не выделяет мать; диффузная мышечная гипотония. По данным МРТ головного мозга обнаружены микроцефалия, истончение мозолистого тела, атрофия лобных долей без кортикального повреждения. В комплекс МАР входили следующие признаки: широкое лицо, эпикант, «голубые» склеры, микростомия, открытый рот, короткая шея, низко расположенные крупные ушные раковины, брахицефалия; двусторонний крипторхизм, гипоплазия гениталий. У ребёнка наблюдались частые респираторные инфекции, пневмонии.
Методом молекулярного кариотипирования была обнаружена терминальная дупликация Хq27.3q28 (рис. 1а) протяженностью 10,6 млн пн, затронувшая 155 генов, 22 из которых индексированы в OMIM (Online Mendelian Inheritance in Man), в том числе гены MECP2, FMR1-AS, FMR1-NB, AFF2, IDS, SLC6A8, BCAP31, L1CAM, AVPR2, FLNA, ATP6AP1, G6PD, F8. Симптомокомплекс пробанда был обусловлен в большей степени дупликацией гена MECP2, известной, как синдром дупликации гена MECP2 [13, 14], выявляемый, в основном, у мальчиков [OMIM:300260].
При «обратном» (повторном) кариотипировании пробанда дополнительный хромосомный материал был обнаружен на длинном плече хромосомы Y, видимый при G- и С- окрашивании (рис. 1б). Последующее FISH исследование с ДНК пробой на участок Xq28, где локализован ген MECP2, показало, что он является материалом хромосомы Х (рис. 1в). Кариотип пробанда – 46,X,der(Y)t(X;Y)(q27.3;q12). При цитогенетическом исследовании, проведенном отцу, выявлен нормальный кариотип – 46,ХY при наличии нестабильности хромосом, кариотип матери – 46,XX. На рисунке видна хромосома Y отца без изменений (рис. 1б).
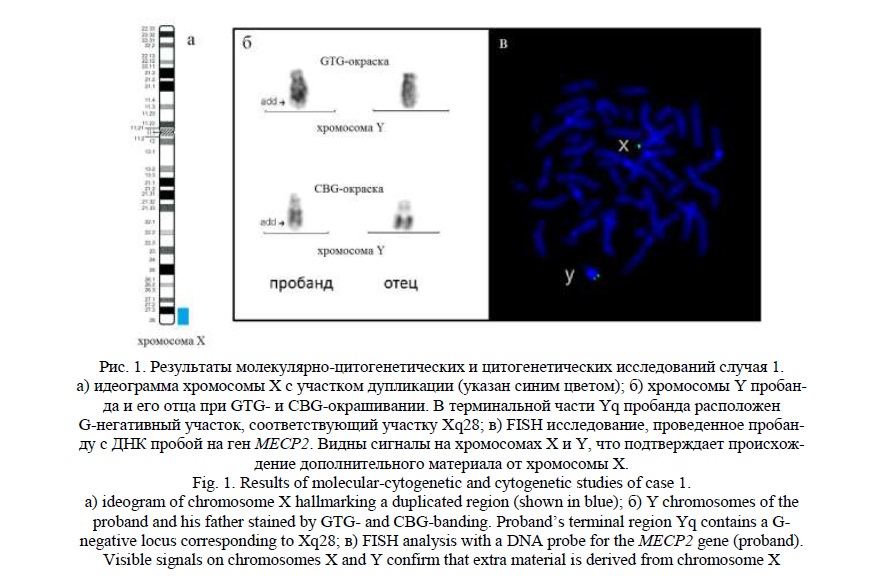
Случай 2.
Клинические признаки у девочки в возрасте 5 лет были следующие: ЗПРР, ЗПМР, микроцефалия; МАР: высокий лоб, гипертелоризм глазных щелей и сосков, врожденная глаукома, широкое переносье, оттопыренные ушные раковины, клювовидный нос, постоянно открытый рот.
Методом молекулярного кариотипирования была обнаружена интерстициальная делеция 2q22.1q22.3 (рис. 2а) протяженностью 7,9 млн пн, затронувшая 63 гена, 6 из которых индексированы в OMIM: NХРН2, LRР1В, КINU, АRНGAP15, GTDC1 и ген ZЕВ2, который ассоциирован с синдромом Моват-Уилсона [15, 16], характеризующимся умственной отсталостью, лицевыми МАР и пороками развития внутренних органов [OMIM:235730].
«Обратным» кариотипированием было обнаружено уменьшение яркости полосы 2q22 у пробанда, что соответствует выявленной делеции (рис. 2б). Кариотип пробанда – 46,ХХ,del(2)(q22.1q22.3). Кариотипы родителей были нормальные (46,ХХ и 46,XY).
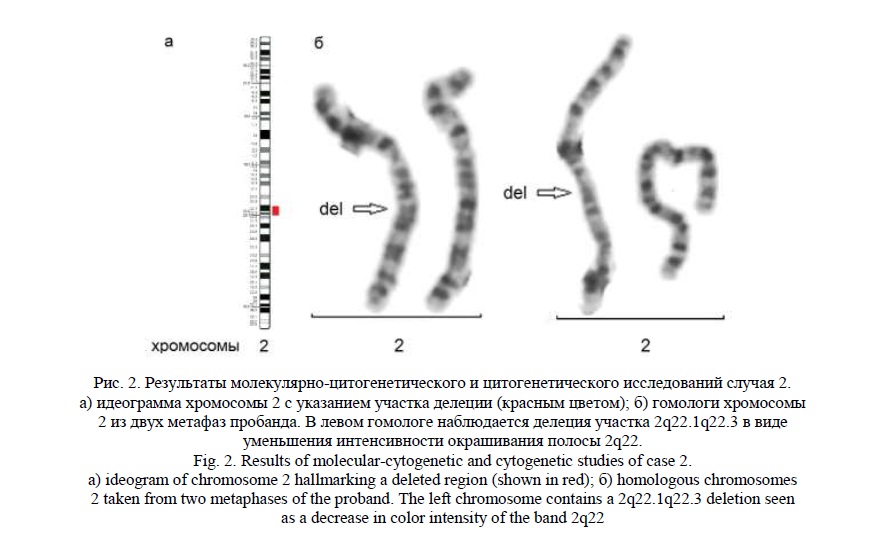
Случай 3.
У девочки в возрасте 2 лет обнаружены следующие клинические проявления: ЗПМР, ЗПРР, нарушение походки, микроцефалия, мышечная гипотония, расщелина твердого и мягкого нёба, миопия, тугоухость 1-й степени, ВПС: открытый аортальный проток, дефекты межпредсердной и межжелудочковой перегородок.
Методом молекулярного кариотипирования была обнаружена интерстициальная делеция 2q22.3q24.1 (рис. 3а) протяженностью 6,7 млн пн, затронувшая 53 гена, 18 из которых индексированы в OMIM: ACVR2A, ORC4, MBD5, EPC2, KIF5C, LYPD6, MMADHC, RND3, NMI, TNFAIP6, RIF1, NEB, ARL5A, CACNB4, STAM2, PRPF40A, RPRM, GALNT13. Делеции в данном геномном участке ассоциированы с умственной отсталостью, аутистическими расстройствами и врожденными пороками развития, в том числе ген MBD5 [17], ассоциированный с синдромом микроделеции 2q23.1, имеющим название «синдром псевдо-Ангельмана» [18] из-за схожих фенотипических проявлений.
«Обратное» кариотипирование выявило сложную интерстициальную структурную перестройку, сочетающую делецию с инверсией (рис. 3б). Кариотип пробанда – 46,ХХ,del(2)(q22.3q24.1),inv(2)(q21.1q22.3). Кариотипы родителей были нормальными (46,ХХ и 46,XY).
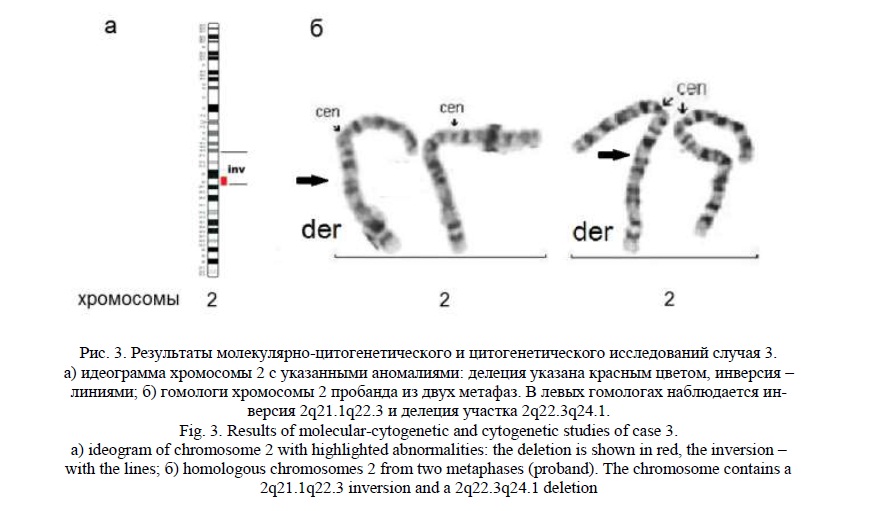
Случай 4.
У девочки в возрасте 1,5 года обнаружены следующие клинические признаки: ЗПМР, задержка физического развитиия (ЗФР), нарушение равновесия, нарушение глотания, порок развития костей черепа (синостоз сагиттального шва), врожденная аномалия дисков зрительных нервов (мегалодиск), лицевые МАР.
Методом молекулярного кариотипирования была выявлена интерстициальная делеция 10q22.1q22.3 (рис. 4а) протяженностью 4,7 млн пн, затронувшая 48 генов, 31 из которых индексирован в OMIM, в том числе следующие гены: DDIT4, DNAJB12, MICU1, MCU, OIT3, PLA2G12B, P4HA1, NUDT13, ECD, DNAJC9, MRPS16, ANXA7, PPP3CB, PLAU,VCL, KAT6B, VDAC2, ZNF503, KCNMA1 .Следует отметить, что гены PLAU, KAT6B, KCNMA1 [19] ассоциированы с задержкой умственного развития, аномалиями головного мозга, лицевыми МАР.
При «обратном» кариотипировании данная делеция визуализировалась под микроскопом при увеличении х1150 благодаря тому, что она захватывала четко различимую полосу хромосомы 10 при дифференциальном окрашивании (10q22.2), отсутствующую в одном из гомологов (рис. 4б). Кариотип пробанда был записан, как 46,ХХ,del(10)(q22q22). Цитогенетическое исследование, проведенное родителям, аномалий хромосом не выявило (кариотипы – 46,ХХ и 46,XY). На рисунке видны гомологи хромосомы 10 родителей без изменений (рис. 4в).
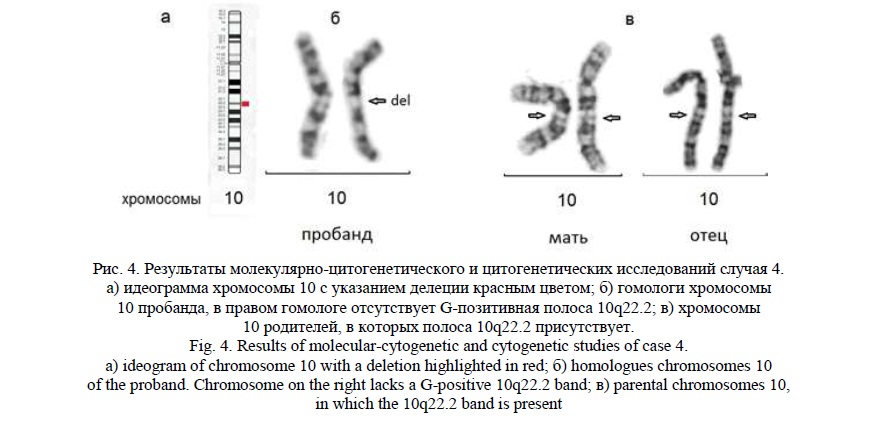
При исследовании методом молекулярного кариотипирования встречаются случаи сочетания терминальных делеций и дупликаций в разных хромосомах. Такое сочетание предполагает наличие несбалансированной транслокации с присутствием делетированной дериватной хромосомы, терминальный участок которой заменен на участок другой хромосомы. Если при этом имеется различие длины фрагментов, участвующих в перестройке, и это различие составляет более 5 млн пн, то цитогенетическое определение перестройки хромосом возможно с большой вероятностью. Но если участки делеции и дупликации имеют равные размеры, выявить такие перестройки цитогенетическим методом весьма сложно; при этом следует обращать внимание на дифференциальное окрашивание перестроенного участка дериватной хромосомы. Ниже приводятся подобные случаи.
Случай 5.
У мальчика в возрасте 4-х лет обнаружены следующие клинические проявления: ЗПРР, ЗПМР, башенная форма черепа, мышечная гипотония, эпилепсия. МАР: телекант, эпикант, широкая переносица, диспластичные низко расположенные ушные раковины, микрогнатия.
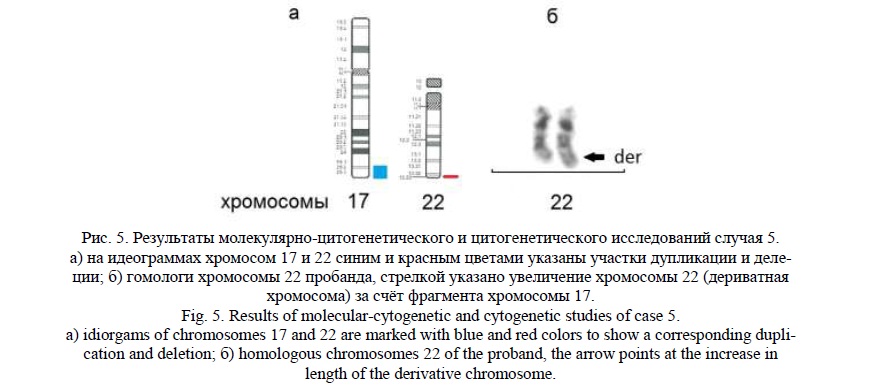
Методом молекулярного кариотипирования были выявлены терминальная делеция 22q13.33 (рис.5а) протяженностью 1млн пн, затронувшая 53 гена, 16 из которых индексированы в OMIM, и терминальная дупликация 17q25.2q25.3 (рис.5а) протяженностью 5,9 млн пн, затронувшая 193 гена, 67 из которых индексированы в OMIM. Наиболее значимым в делетированном участке хромосомы 22 можно выделить ген SHANK3 [20, 21], делеция которого ассоциирована с синдромом Фелан-МакДермид (Phelan-McDermid syndrome, OMIM:606232) [22]. Гены, локализованные в участке дупликации хромосомы 17, ассоциированы с нарушением речевого и психомоторного развития, эпилепсией. В частности, мутации в гене TBCD [23] связаны с прогрессирующей энцефалопатией, атрофией коры головного мозга и истончением мозолистого тела.
Данные результаты указывали на вероятную несбалансированную транслокацию между хромосомами 17 и 22 и наличие у ребенка дериватной хромосомы 22 (рис.5б). Учитывая разницу размеров делеции и дупликации (4,9 млн пн), определенную молекулярным кариотипированием, «обратное» кариотипирование позволило обнаружить увеличение длинного плеча в одной из хромосом 22 (рис. 5а,б). Кариотип пробанда – 46,XY,der(22)t(17;22)(q25.2;13.33). Родители информированы о необходимости проведения цитогенетического и FISH исследований для выявления возможной сбалансированной перестройки хромосом.
Случай 6.
У мальчика в возрасте 2 лет обнаружены следующие клинические проявления: грубая ЗПМР, ЗПРР и ЗФР, микроцефалия, мышечная гипотония; врождённый порок сердца; эпилепсия; МАР: гипертелоризм глазных щелей, эпикант, клювовидный нос, маленький рот с опущенными углами, деформированные низко расположенные ушные раковины. Фенотип напоминал синдром Вольфа-Хиршхорна [24].
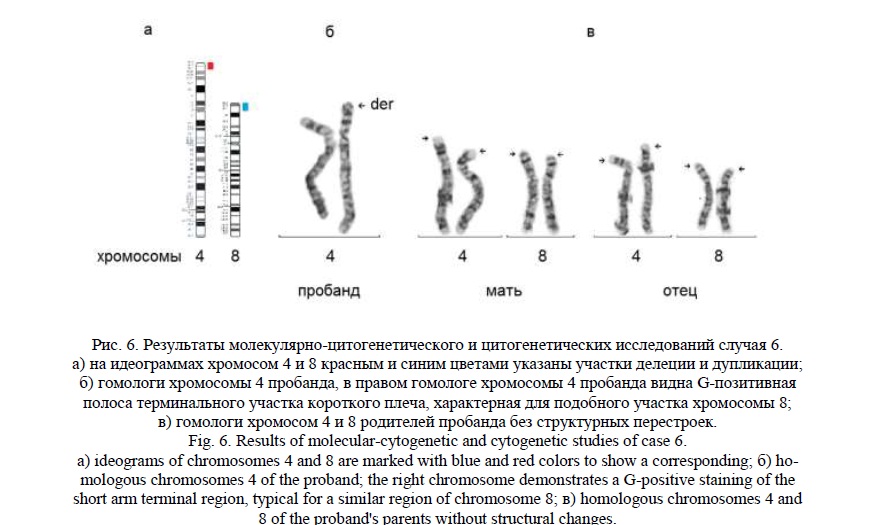
Методом молекулярного кариотипирования были выявлены: терминальная делеция 4р16.3р16.1 (рис. 6а) протяженностью 8,5 млн пн, затронувшая 215 генов, 83 из которых индексированы в OMIM, и терминальная дупликация 8р23.3р23.1 (рис. 6а) протяженностью 6 млн пн, затронувшая 56 генов, 8 из которых индексированы в OMIM. Фенотипические проявления синдрома Вольфа-Хиршхорна могут быть связаны с делецией генов PIGG, CPLX1, CTBP1, LETM1, NSD2 (WHSC1), NELFA (WHSC2), HTT, DOK7, ADRA2C, MSX1, EVC2, MYL5, GAK, C4ORF48 [25, 26]. В участке дупликации хромосомы 8 можно выделить гены CLN8, MCPH1, ARHGEF10 [27, 28], связанные с микроцефалией, умственной отсталостью и эпилепсией. По данным литературы, транслокация между хромосомами 4 и 8 относится к рекуррентным реципрокным транслокациям и занимает второе место по частоте после транслокации между хромосомами 11 и 22 – t(11;22)(q23.3;q11.2) [29, 30].
«Обратное» кариотипирование выявило у пробанда G-позитивную окраску терминальной области 4р, характерную для хромосомы 8 (рис. 6б). Произошла замена G-негативного фрагмента хромосомы 4 на схожий по размеру, но G-позитивный фрагмент хромосомы 8. FISH исследование с ДНК пробой на субтеломерный участок короткого плеча хромосомы 4 показало его отсутствие в кариотипе у пробанда. Кариотип пробанда был записан, как 46,XY,der(4)t(4;8)(p16.1;p23.1). Кариотипы родителей были нормальными (46,ХХ и 46,XY). На рисунке видны гомологи хромосом 4 и 8 родителей без изменений (рис. 6в).
Случай 7.
У девочки в возрасте 7 лет обнаружены следующие клинические проявления: грубая ЗПРР, микроцефалия, эпилепсия; МАР: короткие глазные щели, эпикант, деформация ушных раковин по типу «ухо сатира», широкая переносица, гипоплазия крыльев носа, вывернутые вперед ноздри, глубокий фильтр, широкий рот.
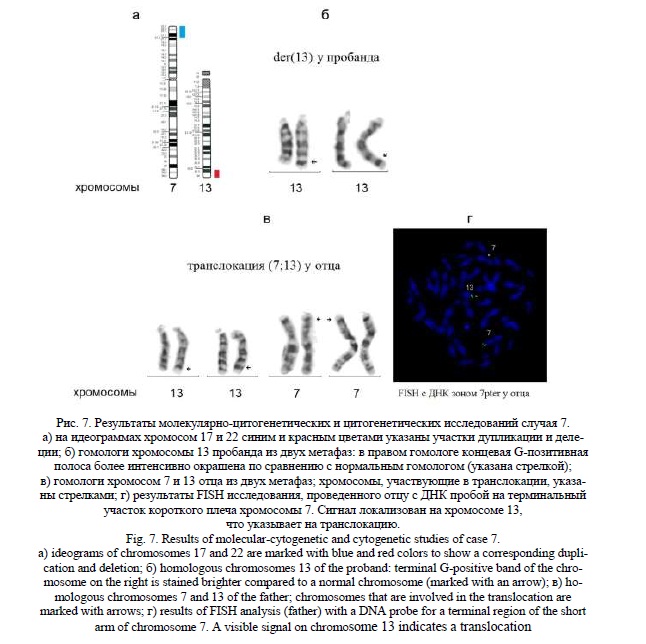
Методом молекулярного кариотипирования были выявлены: терминальная дупликация 7p22.3p21.2 (рис. 7а) протяженностью 13,8 млн пн, затронувшая 102 гена, 58 из которых индексированы в OMIM, и терминальная делеция 13q33.3q34 (рис. 7а) протяженностью 7,5 млн пн, затронувшая 48 генов, 25 из которых индексированы в OMIM. Среди генов, связанных с фенотипическими проявлениями у пробанда, можно выделить следующие: в хромосоме 7 – гены BRAT1, FAM20C, LFNG, CARD11, AP5Z1, ACTB, RNF216, WIPI2, RAC1, ACTB; в хромосоме 13 – LIG4, COL4A1, COL4A2, CARS2, CHAMP1.
Результаты молекулярно-цитогенетического исследования предполагали наличие у ребенка дериватной хромосомы 13 от транслокации с хромосомой 7. «Обратное» кариотипирование выявило усиление интенсивности окрашивания G-позитивной терминальной полосы в длинном плече хромосомы 13, сравнимое по интенсивности с полосой 7р21 (рис. 7б). При кариотипировании родителей была обнаружена сбалансированная транслокация с участием хромосом 7 и 13 (гомологи этих хромосом представлены на рисунке) у отца ребенка (рис. 7в). Для уточнения наличия транслокации отцу было проведено FISH исследование с ДНК пробой на терминальный участок 7р. Сигнал был локализован на хромосоме 13 (рис. 7г). Кариотип пробанда – 46,ХХ,der(13)t(7;13)(p21.2;q33.3), кариотип отца – 46,XY,t(7;13)(p21.2;q33.3).
Случай 8.
У девочки в возрасте 6 лет обнаружены следующие клинические проявления: ЗПРР, ЗПМР, высокорослость; МАР: удлиненная форма лица, частичный птоз, эпикант, широкая переносица, сглаженный фильтр, тонкая верхняя губа, прогнатизм, тонкие оттопыренные ушные раковины со сглаженным рисунком завитка, длинные пальцы кистей и стоп, широкое пупочное кольцо; эпиактивность на ЭЭГ.
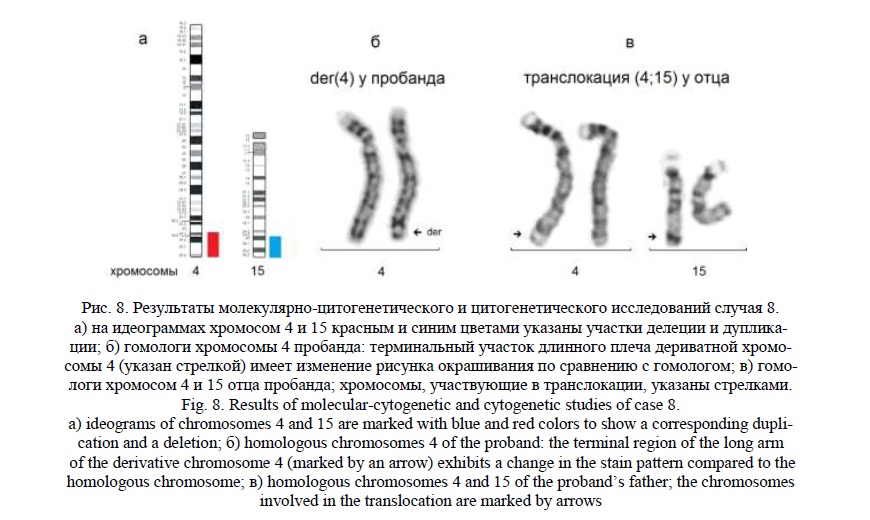
Методом молекулярного кариотипирования были обнаружены: терминальная делеция 4q34.1q35.2 (рис. 8а) протяженностью 14,9 млн пн, затронувшая 202 гена, 35 из которых индексированы в OMIM, и терминальная дупликация 15q25.3q26.3 (рис. 8а) протяженностью 13,6 млн пн, затронувшая 263 гена, 57 из которых индексированы в OMIM. Наиболее значимыми генами в участках перестроек были следующие: для хромосомы 4 – гены TENM3, TRAPPC11, SLC25A4, UFSP2, TLR3; для хромосомы 15 – ACAN, KIF7, LINS1, CHSY1, CHD2. Эти результаты предполагали наличие у ребёнка дериватной хромосомы 4 от транслокации с хромосомой 15, при которой длина дериватной хромосомы практически не изменена, поскольку фрагменты имеют сходные размеры.
«Обратное» кариотипирование выявило изменение рисунка терминальной части 4q (рис. 8б) у пробанда при сохранении длины дериватной хромосомы. При кариотипировании родителей была обнаружена сбалансированная транслокация с участием хромосом 4 и 15 (гомологи этих хромосом представлены на рисунке) у отца ребенка (рис. 8в). Кариотип пробанда был записан как 46,XX,der(4)t(4;15)(q34.1;q25.3), кариотип отца – 46,XY,t(4;15)(q34.1;q25.3), кариотип матери – 46,ХХ.
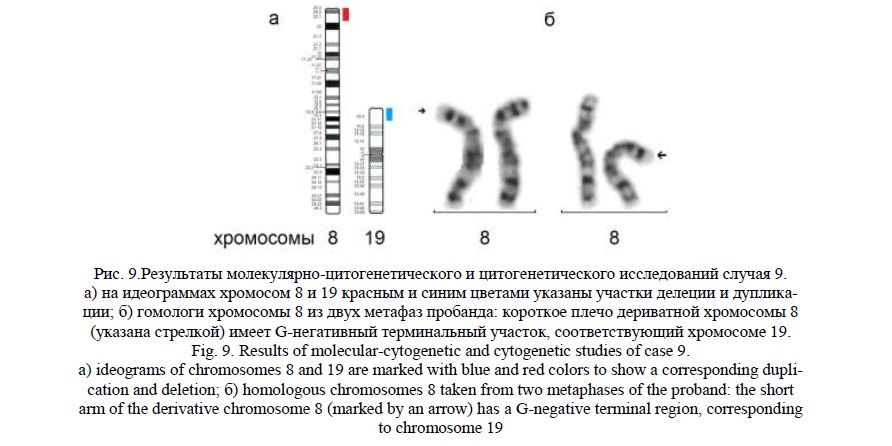
Случай 9.
У мальчика в возрасте 14 лет обнаружены следующие клинические признаки: глубокая умственная отсталость, эпилепсия, атаксия, частичная атрофия зрительного нерва, множественные клиновидные деформации позвонков грудного и поясничного отделов позвоночника, крипторхизм (прооперированный), гинекомастия, гипоспадия, дисплазия тазобедренных суставов, иммунодефицит; МАР: глубоко запавшие глаза, гиперплазия верхней челюсти и десен, диастемы и тремы верхнего зубного ряда, высокое узкое небо, арахнодактилия.
Молекулярное кариотипирование выявило терминальную делецию 8p23.3p23.1 (рис. 9а) протяженностью 7,9млн пн, затронувшую 171 ген, 20 из которых индексированы в OMIM, и терминальную дупликацию19р13.3р13.2 (рис. 9а) протяженностью 7 млн пн, затронувшую 301 ген, 225 из которых индексированы в OMIM. В участке делеции хромосомы 8 можно выделить гены CLN8, MCPH1, ARHGEF10; в дуплицированном участке хромосомы 19 – ADAT3, ABCA7, APC2, AP2D1, LMNB2, TLE6, PIP5K1C, ATCAY, EEF2, TICAM1, CLPP, TUBB4A. Перечисленные гены связаны с умственной отсталостью, эпилепсией, микроцефалией, атрофией коры головного мозга, скелетными аномалиями, атаксией.
Эти результаты предполагают наличие у ребёнка дериватной хромосомы 8 от транслокации с хромосомой 19, при которой длина дериватной хромосомы не изменена, поскольку транслоцированные фрагменты одинаковы по размеру.
При «обратном» кариотипировании пробанда было обнаружено изменение окрашивания терминального участка короткого плеча хромосомы 8 (рис. 9б). Кариотип пробанда был записан как 46,XY,der(8)t(8;19)(p23.1;p13.2). Родители информированы о необходимости цитогенетического обследования.
В эпоху внедрения высокоразрешающих молекулярно-цитогенетических методов исследования, таких как серийная сравнительная геномная гибридизация или молекулярное кариотипирование (arrayCGH и SNParray), встает вопрос об эффективности и целесообразности стандартного кариотипирования. Следует отметить, что метод молекулярного кариотипирования имеет разрешающую способность, превосходящую цитогенетический метод во много раз (1000 пн против 5-7 млн пн). Однако молекулярно-цитогенетические методы, в том числе и молекулярное кариотипирование, способны выявить геномный дисбаланс, тогда как сбалансированные хромосомные перестройки, в основном, определяются цитогенетическим методом (или методом FISH). Кроме того, помимо сбалансированных перестроек, он позволяет выявлять такие изменения генома, как мозаицизм низкого уровня (менее 20% клеток), хромосомную нестабильность, хромосомные онкомаркеры, а также локализацию дуплицированного материала в геноме (например, случай 1) [5, 8, 31].
Выявление носительства сбалансированной перестройки в семье при определённых обстоятельствах является необходимой задачей медико-генетического консультирования [32, 33]. Большое количество генетических повреждений, выявляемых высокоразрешающими методами у больных детей, ставит непростую задачу по обследованию их родителей для прогноза будущего потомства [34, 35, 36]. «Обратное» кариотипирование позволяет частично решить эту задачу путем «таргетного» цитогенетического выявления сбалансированных транслокаций небольшого размера у родителей ребёнка, имеющего несбалансированную перестройку хромосом. Представленные случаи относятся к сложным, «скрытым» структурным хромосомным аномалиям, выявление которых цитогенетическим методом затруднено. В большинстве это уникальные хромосомные перестройки. В случаях, которые мы представляем в данном исследовании, метод молекулярного кариотипирования, эффективно определивший аномалию, заставляет провести повторное «таргетное» цитогенетическое исследование – «обратное» кариотипирование, необходимое для дальнейшего обследования родителей, а при необходимости и других членов семьи. При наличии хромосом высокого разрешения (500-800 полос), как представлено в работе, такая диагностика возможна [3]. Приведенные случаи показывают, что при повторном цитогенетическом исследовании необходимо обращать внимание на изменение рисунка и интенсивности окрашивания полос хромосом, особенно в терминальных участках, для выявления несбалансированных транслокаций, даже в тех случаях, когда длина хромосомы не изменена.
Таким образом, молекулярно-цитогенетические и цитогенетические методы исследования должны использоваться совместно для достижения наиболее корректных результатов в генетической диагностике хромосомных/геномных заболеваний [34, 36].
Заключение. С внедрением высокоразрешающих молекулярно-цитогенетических методов исследования в клиническую практику, таких как серийная сравнительная геномная гибридизация или молекулярное кариотипирование, встает вопрос об эффективности и целесообразности стандартного кариотипирования. Следует отметить, что молекулярно-цитогенетические методы, в том числе и молекулярное кариотипирование, способны выявить геномный дисбаланс, тогда как сбалансированные хромосомные перестройки, в основном, определяются цитогенетическими методами или методом FISH. Кроме того, поскольку цитогенетический анализ проводится на клеточном уровне, помимо сбалансированных перестроек, он позволяет выявлять такие изменения генома, как мозаицизм низкого уровня (менее 20% клеток), хромосомную нестабильность, хромосомные онкомаркеры, а также локализацию аномалий в геноме. «Обратное» (повторное) кариотипирование после применения высокоразрешающего молекулярно-цитогенетического метода определяет аномальную хромосому и её участок «таргетно», исходя из результатов молекулярного исследования. Таким образом, молекулярно-цитогенетические и цитогенетические методы исследования, позволяют наиболее эффективно выявлять различные, как несбалансированные, так и сбалансированные геномные нарушения, что способствует корректной диагностике и эффективности медико-генетического консультирования. Молекулярно-цитогенетические и цитогенетические методы исследования, включая повторное «обратное» кариотипирование, должны использоваться совместно для достижения наиболее корректных результатов в генетической диагностике хромосомных/геномных заболеваний у ребёнка и в его семье.
В отношении данной статьи не было зарегистрировано конфликта интересов.
Благодарности
Исследование частично выполнено в рамках государственного задания № АААА-А18-118051590122-7, «Персонифицированная геномика недифференцированных форм умственной отсталости у детей», 2018-2020 гг.













Список литературы