
Study of microbiome aberrations in patients with irritable bowel syndrome with diarrhea by next-generation sequencing
Aннотация
Background: Irritable bowel syndrome (IBS) is a common functional disorder of the gastrointestinal tract (GIT). Despite the prevalence of this disease, our understanding of the etiology remains limited. However, there is no doubt that microbial factors play a key role in the pathophysiology of IBS. The aim of the study: To investigate the microbiological composition of the intestines of patients with IBS-D. Materials and methods: We used the next-generation sequencing (NGS) method, aimed at targeted sequencing of the hypervariable region V3 of the 16S rRNA gene, which allowed us to study in detail the changes in the microbiome composition in patients. Results: An increase in the number of members of the genera Streptococcus and Haemophilus was revealed in the group of patients with IBS-D, they are mainly represented by opportunistic pathogens and are associated with the development of IBS. At the same time, there was a decrease in bacterial genera: Ruminococcaceae NK4A214, Butyricimonas, Christensenellaceae R-7, Ruminococcaceae DTU089, Coprobacter, Enterococcus and Sciscionella compared to the healthy group. Significant results were also obtained in the correlation analysis between bacteria, showing the relationship of bacteria associated with IBS. Conclusion: Genetic analysis revealed significant differences between the groups of healthy and IBS patients in the composition of the bacterial microbiome. Our results show that the composition of the gut microbiome changes in people with IBS. Thus, this study contributes to understanding the development of IBS-D in terms of microbiome changes.
К сожалению, текст статьи доступен только на Английском
Introduction. IBS is a common functional disorder of the GIT, and it is now clear that this pathology is associated with immune activation and changes in the composition and functions of the intestinal microbiota and the intestinal mucosal barrier [1]. According to studies, the amount of the population worldwide suffering from IBS ranges from 9% to 23%, while the pathophysiology of this disease is still not fully understood [2]. IBS significantly affects the quality of a patient’s life and causes several problems in diagnosis and treatment [3]. The diagnosis of IBS is not confirmed by specific tests or structural abnormalities. The Rome IV criteria are currently the gold standard for IBS diagnosing [4]. The use of intestinal biomarkers in clinical practice can become critical to the precise diagnosis of IBS. Gut microbial biomarkers have been reported to have great potential for detecting inflammatory bowel disease (IBD), colorectal cancer, and autoimmune hepatitis [5].
There are three types of IBS depending on the predominant symptom: diarrhea-predominant (IBS-D), constipation-predominant (IBS-C) and IBS with mixed symptoms of both constipation and diarrhea (IBS-M) [6]. In all appearances, IBS-D is the most common subtype, affecting approximately 40% of patients [7]. Emerging evidence suggests that abnormal changes in the gut microbiota are strongly associated with IBS-D [8]. Despite a large number of studies, there is still no universal algorithm for IBS-D treatment [9]. The diagnosis of IBS-D is primarily based on clinical characteristics, and there are no objective diagnostic tests or validated biomarkers for the diagnosis of IBS-D [10].
The aim of the study. Our research is aimed to study the microbiological composition of the intestines of patients with IBS-D using 16S rRNA gene sequencing on the Ion Torrent Personal Genome Machine (PGM) platform. This work contributes to a better understanding of clinical features and interactions of the IBS-D microbiome and also highlights the potential applications of bacterial biomarkers in the early diagnosis of IBS.
Materials and methods. A total of 20 patients took part in the study. Ten patients were diagnosed with IBS-D based on a complex of complaints, anamnesis data and clinical symptoms (classified using the Rome IV criteria (2016) for irritable bowel syndrome), as well as laboratory diagnostic data, including the following methods: clinical and biochemical blood tests, determination of antibodies to tissue transglutaminase, gliadin, IgA, IgG in blood serum, determination of the level of thyroid hormones, fecal analysis to detect latent blood, fecal analysis to detect bacteria of the intestinal group (Shigella spp., Salmonella spp., Yersinia spp.); toxins A and B of Clostridium difficile; coprogram; analysis of faeces for fecal calprotectin, as well as instrumental research methods: hydrogen breath test with lactulose; ultrasound examination of the abdominal organs. Women were additionally consulted by a gynecologist, according to the indications of pelvic organs ultrasound examination. Ten healthy people without gastrointestinal symptoms were selected with the same method. The selection of patients was carried out at the Olympus of Health Clinic (Voronezh, Russia). All participants gave written consent to the use of their anonymized personal data for research purposes before the examination at the clinic. All procedures were conducted by the ethical standards of the responsible committee on human experimentation (institutional and national) and the Helsinki Declaration of 1964 and later versions. Informed consent or a substitute was obtained from all patients for their participation in the study. The experiment was carried out by the recommendations of the Biomedical Research Ethics Committee of Voronezh State University (protocol No. 42-03 of 10/11/2021).
Fecal samples were obtained from all patients, each sample (approximately 1 gram) was collected into an Eppendorf tube using disinfected plastic equipment after defecation. The samples were immediately cooled and transported in compliance with temperature control to the laboratory.
To study the microbiome composition of faeces obtained from participants examination, DNA was isolated from samples using the ZymoBiomics DNA Miniprep Kit (Zymo Research, Los Angeles, CA, USA) according to the protocol. During the DNA extraction step, we added a sample containing Milli-Q water used in the laboratory as a negative control. This sample was further treated in the same way as the test samples and was sequenced and analyzed. This step is necessary to exclude contamination of the studied samples during the bioinformatic analysis of sequencing data, which allows obtaining an accurate picture of the microbiome. To study the microbiome, we chose the hypervariable region V3 of the 16S rRNA gene. We used a pair of universal primers 337F, 518R for targeted amplification of the study area. Amplification was performed using 5×ScreenMix-HS Master Mix kit (Evrogen, Moscow, Russia) under the following temperature conditions: 94 ℃ for 4 min; 37 cycles of 94 ℃ for 30s, 53 ℃ for 30s, and 72 ℃ for 30s; and final elongation at 72 ℃ for 5 min. Then we proceeded to prepare sequencing libraries for the Ion Torrent PGM platform. For this, we used the commercial NEB-Next Fast DNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) according to the manufacturer's instructions. The libraries were barcoded using NEXTflex DNA Barcodes – Ion Torrent-Compatible – 64 adapters (PerkinElmer, Inc., Waltham, MA, USA). The libraries were then purified with MPureXP magnetic particles (Beckman Coulter, Brea, CA, USA) and quality controlled by qPCR using the Ion Torrent Platforms Library Quantification Kit (Kapa Biosystems, Wilmington, MA, USA). Sequencing was performed on the Ion Torrent PGM system using the Ion PGM Hi-Q View OT2 Kit (ThermoFisher Scientific, Madison, WI, USA) and the Ion PGM Hi-Q View Sequencing Kit (ThermoFisher Scientific, Madison, WI, USA) reagents.
Sequences for each sample were obtained in BAM format. For further analysis, we converted the files to the FASTQ format (FileExporter plugin). Raw sequencing data were available from the NCBI BioProject database (BioProjectID: PRJNA 817720). Bioinformatics processing and phylogenetic analysis of the obtained data were performed using the R programming language in the R-studio environment (VSEARCH v.2.8.2 software). All received reads were subjected to the processes of filtering low-quality sequencing reads, as well as trimming the reads to a constant length and demultiplexing. Filtering was carried out using the maximum expected error threshold equal to 1.0 (DADA2 package). At the end of these manipulations, we performed dereplication, which combined all identical sequencing reads into unique sequences. Then we proceeded to search for operational taxonomic units (OTUs). Taxonomy at the genus level was determined with 100% identity with amplicon sequence variants using version 132 of the SILVA database (https://www.arb-silva.de, accessed 30 August 2022). Using the phyloseq package, we combined the obtained phylogenetic data. At this stage, we carried out the identification and subsequent removal of possible laboratory contaminants (decontam package). After that we proceeded to taxonomic filtering, assessment of the prevalence in samples of individual taxa and agglomeration (phyloseq package).
To compare relative abundances between experimental groups, we used the generalized linear modelling (GLM) method implemented in the DeSEQ2 R package. P-values for each OTU are obtained using with Benjamini-Hochberg multiple inference correction. Alpha diversity for each group was calculated with the Shannon and Chao1 indexes. We use the nonparametric Mann-Whitney test and pairwise adjusted (Holm) p-values to assess differences in alpha diversity scores between groups. Beta diversity was represented by Principal Coordinate Analysis (PCoA), and the distance between the microbial compositions of the samples was calculated using Bray-Curtis dissimilarity. The presence of statistically significant differences in microbial composition between the two study groups was assessed using permutation multivariate analysis of variance (PERMANOVA). Diversity visualization was performed using the phyloseq package. The correlation of the genera for each group was statistically analyzed using the Spearman correlation. All the used genera nodes were R > |0.8| and p≤0.05.
Results and discussion. Progress in the development of high-throughput sequencing methods and bioinformatics tools has led to a shift from clinical microbiology to a genomic nature of research. NGS has expanded our understanding of the human microbiome by allowing the detection and characterization of non-culturable microbes [11]. This work included a study of the microbiome profile at the genus level in 20 patients who were divided into two equal groups. As a result of sequencing, we obtained 767362 reads in total, which corresponded to 229 genera. For further analysis, all bacterial genera, the total abundance of which was less than 0.005 for each research group, were combined into Other genus. Figure 1 shows the most common bacterial genera found in patients.

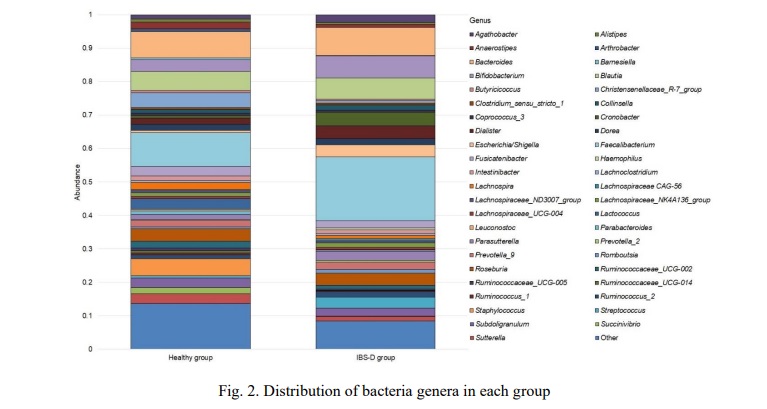
Members of the genus Faecalibacterium predominated in both groups, in the healthy group it was 0.1020±0.0138 while in the disease group it was 0.1907±0.0152. The next largest genus in both cases is Bacteroides 0.0776±0.0194 in the healthy group vs. 0.0844±0.0258 in the IBS-D. The next in the healthy group were Blautia 0.0569±0.0128, Staphylococcus 0.0508±0.0504, Christensenellaceae R-7 group 0.0438±0.0280, Roseburia 0.0375±0.0126, Bifidobacterium 0.0348±0.0174, Lactococcus 0.0310±0.0233, Sutterella 0.0294±0.0155 and Subdoligranulum 0.0284±0.0086. At the same time the IBS-D group the next genus dominated: Bifidobacterium 0.0655±0.0244, Blautia 0.0636±0.0145, Cronobacter 0.0413±0.0262, Dialister 0.0383±0.0111, Escherichia/Shigella 0.0367±0.0273, Roseburia 0.0361±0.0632, Streptococcus 0.0336±0.0148 and Parasutterella 0.0281±0.0095.
A comparative assessment of alpha diversity, calculated using the Shannon and Chao1 indexes, is shown in Figure 3.

There were no statistically significant differences in alpha diversity scores on both tests between groups (Shannon Index, non-parametric Mann-Whitney test with Holm. corrected p=0.32).
Beta diversity is shown by the dissimilarity among samples according to the microbial composition depicted in the PCoA analysis (Bray-Curtis). According to our data the microbial composition of the healthy and the IBS-D groups was not different (Fig. 4).

Analysis of differences in microbiome composition in the IBS-D group compared with the healthy group at the genus level was performed using the DeSEQ2. P-values for each OTU are obtained using with Benjamini-Hochberg multiple inference correction. As a result, statistically significant differences were found for nine genera (Fig.5).

According to the data obtained in the group of IBS-D patients, an increased number of two genus Streptococcus by 2.1586 times (p=0.0002) and Haemophilus by 4.6669 times (p=0.0001) was observed compared with the control. At the same time, in the group of patients, a decrease in the genera Ruminococcaceae NK4A214 group by 2.2899 times (p=0.0019), Butyricimonas by 2.7718 times (p=0.0015), ChristensenellaceaeR-7 group by 2.9933 times (p=0.0004), Ruminococcaceae DTU089 by 3.6969 times (p=0.0002), Coprobacter by 3.7335 times (p=0.0016), Enterococcus by 4.1824 times (p=0.0001) and Sciscionella by 7.3759 times (p=0.0006) was registered compared to the healthy group.
Spearman's correlation analysis using a two-tailed p-value showed that in the healthy group there is a complete correlation (R=1.0, p<0.05) between the genus Kosakonia and the genera Ochrobactrum (p=0.0111) and Pseudochrobactrum (p=0.0111); Sporobacter and Alloscardovia (p=0.0111); Brevundimonas and Phyllobacterium (p=0.0111). In the IBS-D group, we observed a complete correlation between the genera Corynebacterium and Erysipelatoclostridium (p=0.0001); the genus Leucobacter and the genera Polymorphobacter (p=0.0014) and Phyllobacterium (p=0.0014); Polymorphobacter and Phyllobacterium (p=0.0014).
The genus Faecalibacterium, which was characterized by the highest abundance in both groups, directly correlated in the control group with the genera Tyzzerella (R=0.8061, p=0.0072), Peptococcus (R=0.8815, p=0.0014), Ruminococcaceae V9D2013 group (R=0.8283, p=0.0052), Negativibacillus (R=0.8182, p=0.0058), Ruminiclostridium 5 (R=0.8571, p=0.0025), Oscillospira (R=0.8303, p=0.0047), Faecalitalea (R=0.8942, p=0.0011), Anaerofilum (R=0.8598, p=0.0029). In the group of patients with IBS-D, Faecalibacterium was negatively correlated only with the genus Anaerostipes (R=-0.8303, p=0.0047).
The use of NGS methods aimed at targeted sequencing of hypervariable regions of the 16S rRNA gene makes it possible to study in detail changes in the taxonomic composition compared to classical microbiological approaches.
Our study revealed changes in the composition of the gut microbiota in patients with IBS-D. In particular, an increase in the number of bacteria of the genera Streptococcus and Haemophilus was found.
The genus Streptococcus belongs to phylum Firmicutes and consists of 104 recognized species, including both commensal and pathogenic microorganisms [12]. Since the limitation of our research method is the impossibility of identifying bacteria to the species, we cannot reliably state the number of which beneficial or pathogenic members of this genus increased. However, our results are consistent with previous findings that an increase in Streptococcus bacteria may cause IBS symptoms [13-16].
The genus Haemophilus belongs to the family Pasteurellaceae and the class Gammaproteobacteria [17]. Members of this genus are ubiquitous in the human oral cavity, upper respiratory tract and intestines [18]. The genus consists of a variety of species, including some that are pathogenic to humans [19]. It was previously noted that in IBS, the content of bacteria of the genus Haemophilus increases [20, 21, 22]. In our study, we also observed an increase in the number of members of this genus. It can be concluded that most likely these bacteria do play an important role in the development of IBS.
We also observed a decrease in the number of bacteria of some genera, in particular Ruminococcaceae NK4A214 group. Microorganisms belonging to this genus are producers of butyrate [23]. Bacteria that produce short-chain fatty acids (SCFAs) are known to be generally reduced in the faeces of people with IBS compared to healthy people, consistent with our results [24].
We found a decrease in the members of the genus Butyricimonas in patients with IBS-D. Members of this genus are Gram-negative anaerobic bacteria belong to butyrate producers [25]. They are present in the intestinal tract of several mammals, including rats and humans [26]. A hypothesis based on research data suggests that Butyricimonas species have a positive effect on the host's energy metabolism and are also involved in commensal homeostasis between the gut microbiota and the health status of the host. Treatment of metabolic disorders with metformin and statins has been shown to significantly increase the relative abundance of Butyricimonas spp. in the intestine, which significantly correlated with metabolic parameters [27, 28]. However, the metabolic mechanisms by which Butyricimonas exerts metabolic improvements are not understood. According to some studies, bacteria of this genus have therapeutic potential and are used as one of the components of the prevention and treatment of IBS in patients [29]. Thus, our study also showed a positive effect on the human body and an exceptional correlation with IBS-D.
Also, the results of our study indicate the depletion of the genus Christensenellaceae R-7 group in patients with IBS-D. This genus belongs to the family Christensenellaceae, which was recently isolated from the human intestine and shows a strong relationship with the health of the host [30]. The relationship between the high number of the Christensenellaceae R-7 group and normal body mass index (BMI) is known [30, 31]. Christensenellaceae family and the genus Christensenellaceae R-7 group in particular is also known to be depleted in IBD patients such as ulcerative colitis and Crohn’s disease [32]. Several studies have also reported a reduction in the number of Christensenellaceae in IBS, a gastrointestinal disorder characterized by abdominal pain and abnormal bowel movements, compared with healthy individuals. Several studies have noted a positive correlation between Christensenellaceae and longer transit times or even constipation. In our study, we studied the microbiota in patients with IBS-D, which is characterized by diarrhea. Thus, our observations are consistent with the findings that Christensenellaceae is depleted under conditions associated with inflammation and rapid transit [33, 34, 35].
The Ruminococcaceae family has been identified as one of the biomarkers of healthy microbiota [36]. In our study, we found a decrease in the genus Ruminococcaceae DTU-089 in the IBS-D patient group, however, no statistically significant aberrations were observed at the level of the Ruminococcaceae family compared to the healthy group. This genus is poorly researched and its role and mechanisms in the human gut are not fully understood. It is only known that some bacteria Ruminococcaceae DTU-089 can produce methane [37]. The degree of methane production is associated with greater severity of constipation in IBS-C, however, in our study, the type of IBS associated with diarrhea was studied, which may explain the decrease in methane-producing microorganisms, particularly Ruminococcaceae DTU-089 [38]. One study investigating the effects of arabinogalactan on the gut microbiome showed that it modulates the gut microbiome by significantly reducing Firmicutes and increasing Bacteroidetes and Bifidobacterium as well as Ruminococcaceae DTU-089. It can also modulate the metabolic functions of the gut microbiota [37]. Based on the available literature data, further studies of the genus Ruminococcaceae DTU-089 and its functional and metabolic effects in the human intestine are needed.
It is known that the bacterial genus Coprobacter belongs to the family Porphyromonadaceae, the order Bacteroidales and the type Bacteroidetes [38]. It is known that members of the genus Coprobacter are widely distributed in the human intestinal microbiota and are noted as a genus capable of producing propionic acid [39, 40]. In our study, this genus of bacteria showed a decrease in the population with IBS. However, the significance of members of the genus Coprobacter on human health is still not fully known.
Enterococcus is an important pathogen that can damage the intestinal mucosa and thereby cause dysfunction of the immune defenses in patients with IBS [41]. A systematic review of studies by Zhuang and colleagues (2017) found a higher level of Enterococcus in patients with IBS compared with healthy people [42]. However, according to the results of our study, a decrease in the number of this bacterial genus was noted in patients with diagnosed IBS in comparison with the healthy group.
The bacterial genus Sciscionella, first described in 2009, belongs to Gram-positive, aerobes, marine actinomycetes [43]. Data reporting an association of the genus Sciscionella with symptoms of IBS has not been described. According to the results of our study, in the IBS-D group, members of this genus decreased in comparison with the control group. Previously, this bacterial genus was associated with the development of infection in humans [44]. Also, in a study by Sáez-Nieto et al. (2021), the genus Sciscionella was identified in isolates of clinical specimens [45].
Spearman's correlation analysis using a two-tailed p-value showed that in the healthy group there is a complete correlation (R=1.0, p<0.05) between the genus Kosakonia and the genera Ochrobactrum (p=0.0111) and Pseudochrobactrum (p=0.0111); Sporobacter and Alloscardovia (p=0.0111); Brevundimonas and Phyllobacterium (p=0.0111). In the IBS-D group, we observed a complete correlation between the genera Corynebacterium and Erysipelatoclostridium (p=0.0001); the genus Leucobacter and the genera Polymorphobacter (p=0.0014) and Phyllobacterium (p=0.0014); Polymorphobacter and Phyllobacterium (p=0.0014).
The bacterial genera Ochrobactrum and Pseudochrobactrum belong to the family Brucellaceae [46, 47]. These genus shows resistance to antibiotics [48]. The bacterial genus Ochrobactrum is considered opportunistic for humans, producing the development of infection in an organism with a weakened immune system [49].
Members of the genus Alloscardovia, considered to be anaerobic bacteria, are referred to as Bifidobacterium and have been identified in various biological specimens: urine, blood, oral cavity, urethral specimens, tonsil and lung abscess specimen, and aorta [50, 51]. In turn, members of the genus are considered pathogenic for the body associated with infectious diseases [52, 53]. Also associated with infectious diseases is the bacterial genus Brevundimonas [54].
Members of the genus Phyllobacterium are nonpathogenic bacteria that were first isolated from leaf nodules of some plant families [55]. However, a high number of members of Phyllobacterium was noted in gastric carcinoma and cystic fibrosis, as well as in gastric cancer [56, 57, 58].
Members of the genus Corynebacterium are typical human skin bacteria often found in the nasal cavity [59]. Literature data on association with inflammatory processes in the body were not found. It is also known that the genus Leucobacter contains genes encoding glycoside hydrolases and carbohydrate binding modules that are directed to the breakdown of starch and oligosaccharides [60]. Previous studies have noted that patients with gout have an increased number of opportunistic microorganisms of the bacterial genus Erysipelatoclostridium in the fecal microbiome [61]. But literature data confirming the relationship of these births with intestinal health was not found. There are also no data on the association of the bacterial genera Kosakonia, Sporobacter, Polymorphobacter with the intestinal microbiome.
The genus Faecalibacterium, which was characterized by the highest abundance in both groups, directly correlated in the control group with the genera Tyzzerella, Peptococcus, Ruminococcaceae V9D2013 group, Negativibacillus, Ruminiclostridium 5, Oscillospira, Faecalitalea, Anaerofilum. Members of the genera Tyzzerella, Peptococcus and Negativibacillus are normal colonizers of the human intestine, but an increase in their number may be associated with the appearance of various pathologies, including the digestive tract [61-66]. Members of the genus Ruminiclostridium 5 and Oscillospira can produce butyrate and are associated with positive effects on host health, a direct association of abundance with low fat, in addition to solid feedback with IBD [67, 68, 69]. Faecalitalea and Anaerofilum are also potentially beneficial bacterial genera that play an essential role in the pathogenesis of diabetes mellitus and reduce insulin secretion, as well as having an inverse relationship with overweight [70, 71].
In the group of patients with IBS-D, Faecalibacterium was negatively correlated only with the genus Anaerostipes which is considered one of the key intestinal genera associated with human health and disease, as its members are capable of producing butyrate, which is known to have beneficial effects on intestinal functions [72].
Thus, in the group with IBS, we observed an increase in such genera as Streptococcus and Haemophilus, which are mainly represented by opportunistic pathogens and, in accordance with the literature data, are associated with the development of IBS. At the same time, we found a decrease in SCFA-producing bacteria such as Ruminococcaceae NK4A214 group, Butyricimonas and Coprobacter in the group of IBS patients. Significant results were also obtained in the correlation analysis between bacteria, showing the relationship of bacteria associated with IBS. However, our study has a limitation of a relatively small sample size, but this is comparable to sample sizes in similar published studies. Together with previously published data, our results show that the composition of the gut microbiome changes in people with IBS.
Conclusion. We found significant differences between the groups of healthy and IBS patients in the composition of the bacterial microbiome. Thus, this study contributes to understanding the development of IBS-D in terms of microbiome changes, which may lead to the development of new strategies in the preventive diagnosis and treatment of this disease in the future. All this emphasizes the need for further work aimed at studying the microbiome composition and its contribution to the development of IBS-D.
Financial support
This work was supported by the Ministry of Science and Higher Education of the Russian Federation in the framework of the “Science” national project (project FZGW-2020-0001, unique number of the Register of State Tasks 075001X39782002).













Список литературы
Список использованной литературы появится позже.