
Молекулярное кариотипирование хромосомных аномалий и вариаций числа копий последовательностей ДНК (CNVs) при идиопатических формах умственной отсталости и эпилепсии
Aннотация
Актуальность: В этиологии недифференцированных форм умственной отсталости и эпилепсии значимую роль играют генетические факторы. С интенсивным внедрением современных технологий в клиническую практику стало возможным выявлять геномные перестройки с недоступным ранее разрешением. Несмотря на прогресс в изучении генетических причин умственной отсталости, для определения патогенетических механизмов таких этиологически гетерогенных состояний, как её недифференцированные формы с эпилепсией, требуется применение биоинформатических методов для корректной интерпретации результатов полногеномного анализа. Цель исследования: Определение геномных (хромосомных) вариаций, включая CNVs, и их возможных клинических последствий у детей с недифференцированной умственной отсталостью и эпилепсией. Материалы и методы: Цитогенетическими и молекулярными технологиями исследованы клетки крови и образцы ДНК у 294 детей с недифференцированной умственной отсталостью и эпилепсией с ВПР и/или МАР. Использовали молекулярное кариотипирование или SNParray и оригинальную биоинформатическую технологию, позволяющую моделировать последствия геномных аномалий. Результаты: Обследовано 294 ребёнка с геномными аномалиями и различными клиническими проявлениями. В 20,8% случаев помимо идиопатической умственной отсталости, ВПР и/или МАР, наблюдалась эпилепсия. У этих детей обнаружены численные и структурные аномалии хромосом в 8% случаев. При молекулярном кариотипировании выявлено 192 аномалии генома с патогенным или вероятно патогенным эффектом и 23 участка сегментной потери гетерозиготности (унипарентальная дисомия) в 25% случаев. При этом сочетанные аномалии генома наблюдались в 87%. Геномные аномалии встречались по всем хромосомам, кроме 20 и 21. Среди 192 геномных аномалий выявлены делеции, дупликации, трипликации и мозаичные геномные нарушения, как правило, совместно с регулярными перестройками. При применении оригинального биоинформатического анализа, используя приоритизацию генов, определено более 800 генов; из повторяющихся генов выявлены: FMR1 [OMIM:309550], ассоциированный с умственной отсталостью, сцепленной с ломкой хромосомой X; DAZ2 [OMIM:400026] и DAZ3 [OMIM:400027], ассоциированные c умственной отсталостью и аутизмом; BTRC [OMIM603482], вовлеченный в сигнальный путь циркадного ритма, связанного с эпилептическими проявлениями; реже - AFF2 (FMR2) [OMIM:300806], SLC1A1 [OMIM:133550], SCN2A [OMIM:182390], SCN3A [OMIM:182391], GABRB3 [OMIM:137192], NECAP1 [OMIM:615833], SHANK3 [OMIM:606230]. Вариабельность полученных результатов не позволяет провести корректные корреляции геномных нарушений и недифференцированной умственной отсталости с эпилепсией. Однако данные проделанной работы показывают, что следует накапливать результаты полногеномных исследований для определения геномного участка или даже генов, связанных с данной патологией. Заключение: Полученные данные и проведённый анализ указывают на целесообразность продолжения исследований, направленных на поиск молекулярных механизмов недифференцированной умственной отсталости и эпилепсии.
Ключевые слова: недифференцированная умственная отсталость с эпилепсией, молекулярное кариотипирование, вариации числа копий последовательностей ДНК (CNVs), биоинформатическая технология
Введение. Умственная отсталость относится к одной из самых распространенных групп заболеваний, связанных с нарушениями функционирования головного мозга. Распространенность умственной отсталости в разных странах составляет от 1 до 3% в популяции [1, 2, 3]. Этиология устанавливается приблизительно у 35% умственно отсталых детей. Остальные случаи – это идиопатические (недифференцированные) формы. Недифференцированные формы умственной отсталости часто встречаются с эпилепсией [1-4].
Эпилепсия, как известно, это нарушение церебрального характера, связанное с повторяющимися стереотипными припадками, как правило, возникающими без явных провоцирующих факторов вследствие аномальных синхронных вспышек возбуждения нейронов коры головного мозга. Эта хроническая патология головного мозга с периодически повторяющимися судорогами или их эквивалентами встречается у детей с частотой 1,5-5% [2, 5]. Заболевание различно по своему течению: от доброкачественных до тяжёлых «злокачественных» состояний, включая формы с умственной отсталостью, которые могут сопровождаться регрессом в психическом развитии при различной степени инвалидизации. По данным литературы до 35% умственно отсталых детей страдают повышенной эпилептической готовностью [2, 4].
В этиологии и патогенезе недифференцированных форм умственной отсталости и эпилепсии значимую роль играют генетические факторы. Эти факторы ассоциированы с незрелостью головного мозга и с генетически обусловленной нестабильностью мембран нейронов. Они могут быть связаны с наследственными дефектами обмена, митохондриальными нарушениями, геномными (хромосомными) аномалиями и синдромами, в том числе микроделеционными/микродупликационными, а также с такими наследственными болезнями, как туберозный склероз и нейрофиброматоз. Среди геномных нарушений известны «крупные» хромосомные аномалии, вариации числа копий последовательностей ДНК (CNV) и эпигеномные вариации (однородительское происхождение отдельных участков гомологичных хромосом) [1, 6, 7]. Геномные аномалии можно обнаружить с помощью таких методов, как стандартное и молекулярное кариотипирование. Метод стандартного кариотипирования позволяет обнаружить хромосомные аномалии размером 5-8 млн пн и более, тогда как молекулярное кариотипирование является более чувствительным методом и позволяет обнаружить геномные перестройки размером 1000 пн и менее. Согласно данным литературы, в исследованиях, проведённых с помощью метода молекулярного кариотипирования, в группах детей с недифференцированной умственной отсталостью, аутизмом, ВПР и/или МАР у 25% из них обнаружены «крупные» хромосомные (геномные) аномалии размером от 500 тысяч пн, а у более чем половины детей аномалии генома от менее1 до 500 тысяч пн, ассоциированные с психическими нарушениями [8, 9].В группе детей с идиопатической эпилепсией геномные аномалии размером от 13 тысяч до 16 млн пн, которые могли быть связаны с судорогами, встречались в 8,9% случаев[5].Использование современных методов исследования генома, а именно, высокоразрешающего молекулярного кариотипирования (полногеномное сканирование изменений числа копий последовательностей ДНК–CNV) даёт информацию о размере и координатах выявленных нарушений, позволяет успешно идентифицировать гены, затронутые геномными изменениями, а также определять молекулярные и клеточные процессы, экзогенная коррекция которых рассматривается, как одна из наиболее многообещающих форм научно обоснованной терапии. При этом, несмотря на значительный прогресс в изучении генетических причин различных форм генетически обусловленных болезней, для определения патогенетических механизмов таких этиологически гетерогенных состояний, как умственная отсталость с эпилепсией, требуется применение сложной биоинформатической (интерпретационной) технологии insilico для более детальной и корректной интерпретации полученных индивидуальных данных по исследованию генома [7, 10, 11, 12]. В данной статье представлены результаты цитогенетического, молекулярно-цитогенетического, включая биоинформатическую технологию, и клинического исследований детей с недифференцированной формой умственной отсталости и эпилепсии.
Цель исследования. Целью исследования явилось определение геномных вариаций, в том числе хромосомных аномалий и вариации числа копий последовательностей ДНК (CNVs), а также возможных клинических последствий CNVs у детей с недифференцированными формами умственной отсталости и эпилепсией.
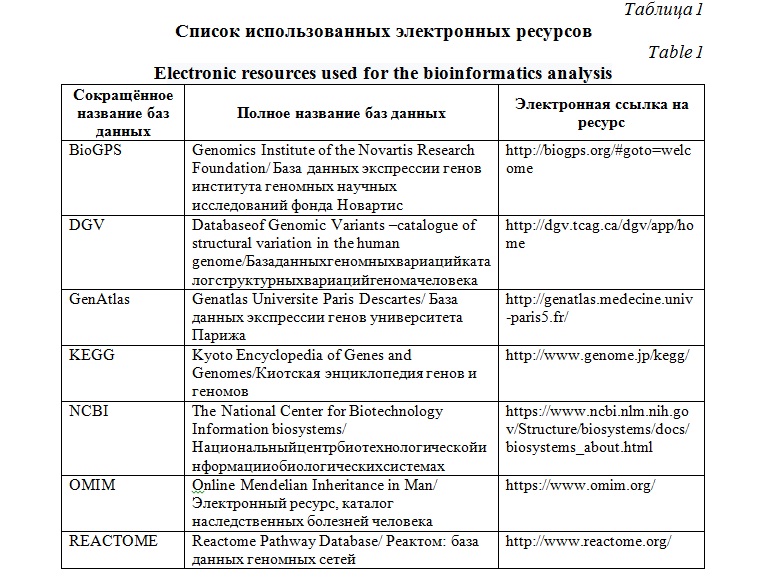
Материалы и методы исследования. Для достижения поставленной цели с помощью цитогенетических и молекулярно-цитогенетических технологий были исследованы клетки лимфоцитов периферической крови и образцы ДНК у294 детей с недифференцированными формами умственной отсталости и эпилепсией, ВПР и/или МАР. Исследования проводились с помощью методов классического цитогенетического кариотипирования и высокоразрешающего молекулярного кариотипирования (полногеномное сканирование изменений числа копий последовательностей ДНК или SNP-arrayс применением микроматриц/чипов AffymetrixCytoscan HD) и оригинальной биоинформатической технологии, позволяющей моделировать последствия геномной патологии [1, 10]. Оригинальная биоинформатическая технология подробно описана в предыдущих публикациях наших лабораторий [11, 12, 13]. Использованные в работе биоинформатические ресурсы и базы данных представлены в таблице 1.
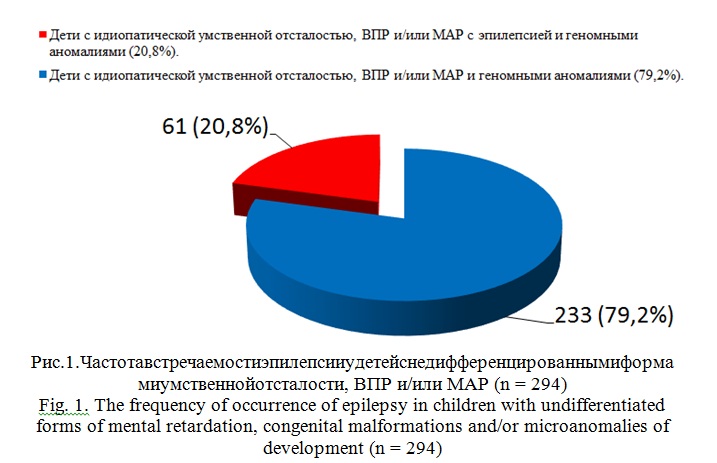
Результаты и их обсуждение. В ходе исследования детей с недифференцированными формами умственной отсталости, ВПР и/или МАР с помощью молекулярного кариотипирования нами было обследовано 294 ребёнка с геномными аномалиями. Среди них, у 61 ребенка, что составило 20,8%, помимо идиопатической умственной отсталости, ВПР и/или МАР, в клинической картине наблюдалась эпилепсия (рис. 1). Полученные результаты соответствуют данным литературы, из которых известно, что эпилепсия у умственно отсталых детей встречаетсяcчастотой от 5,5%до 35% [2, 5].
Результаты геномных и клинических исследований детей с недифференцированными формами умственной отсталости, эпилепсией, ВПР и/или МАР, представлены в таблице 2.

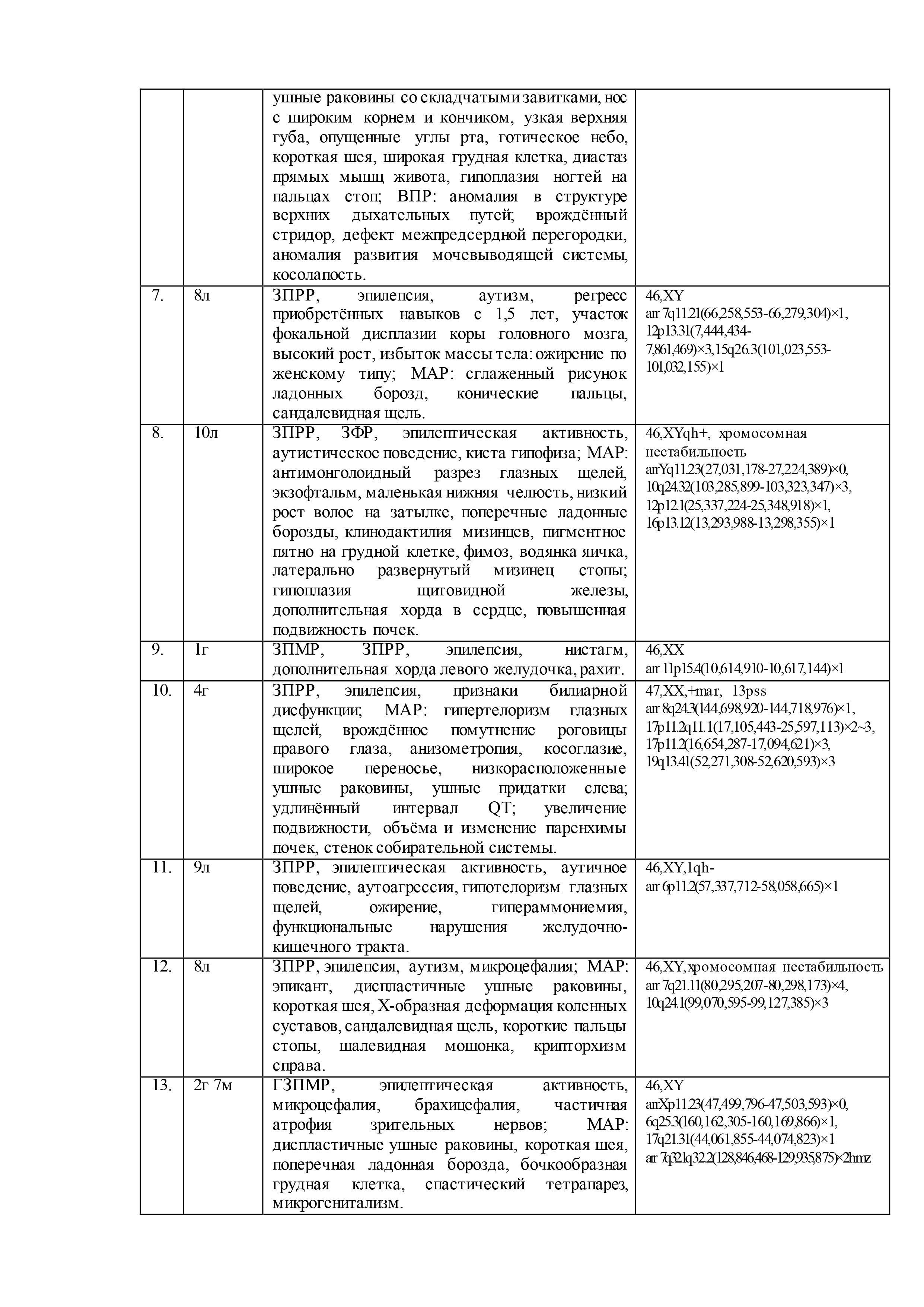
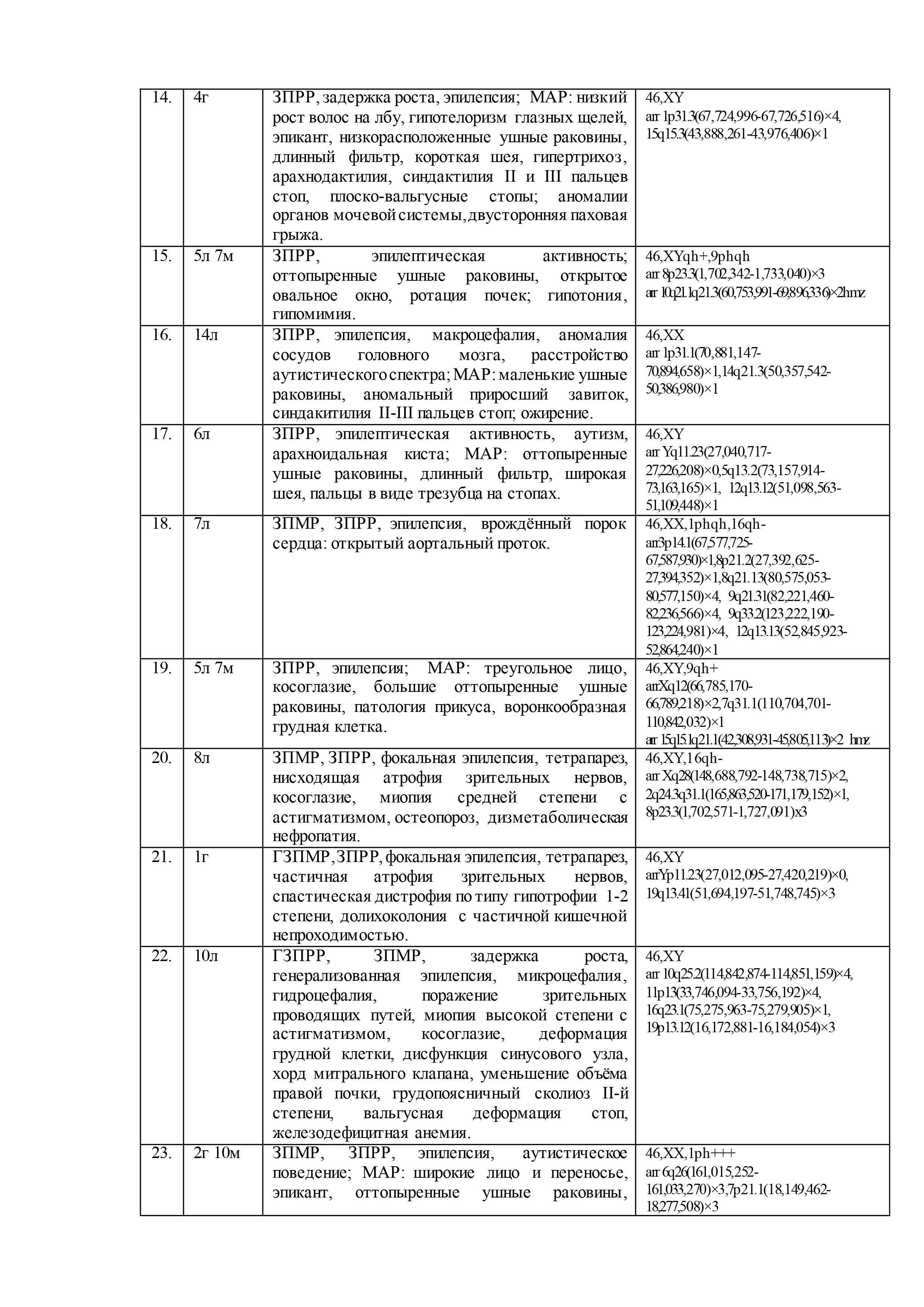
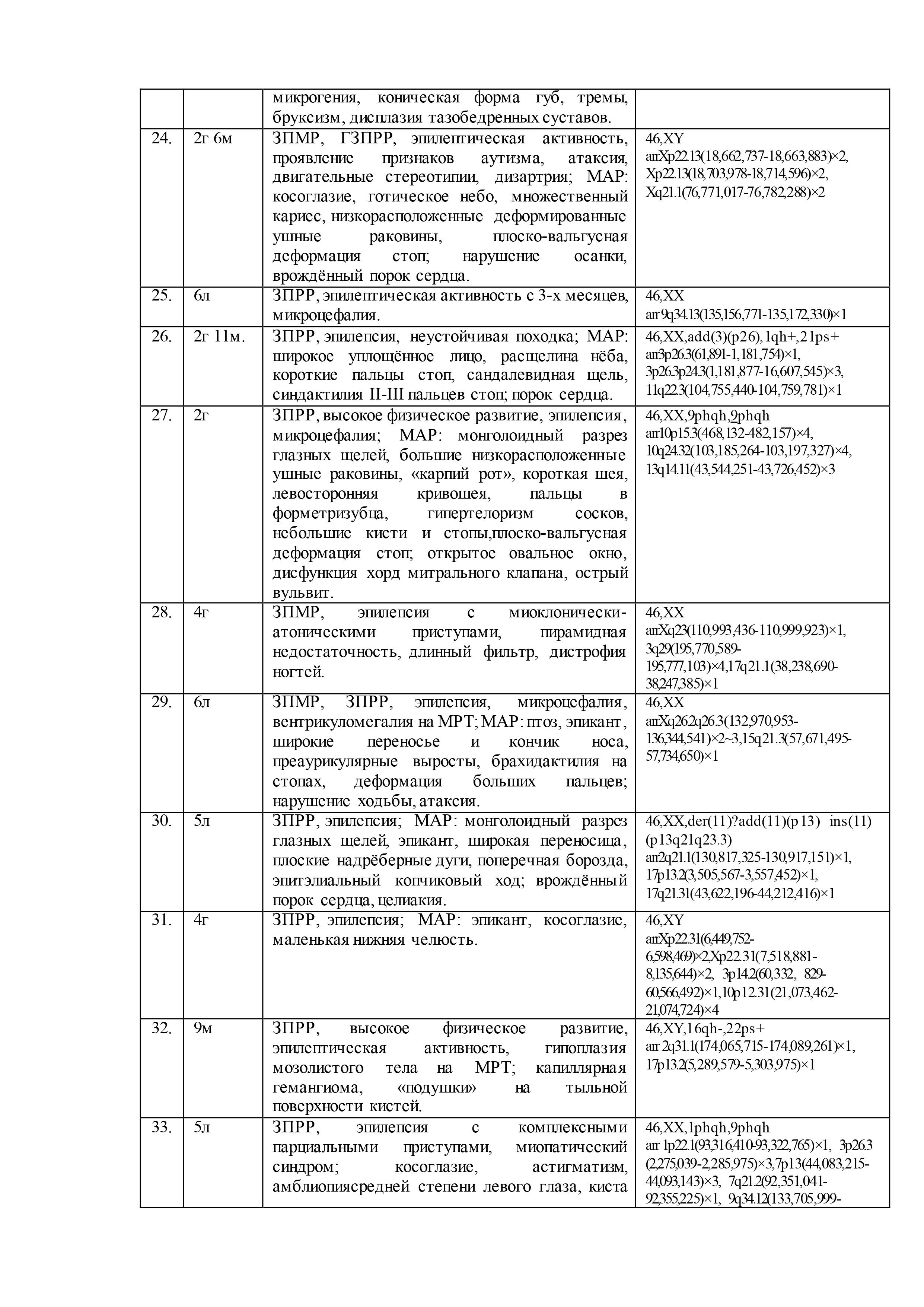
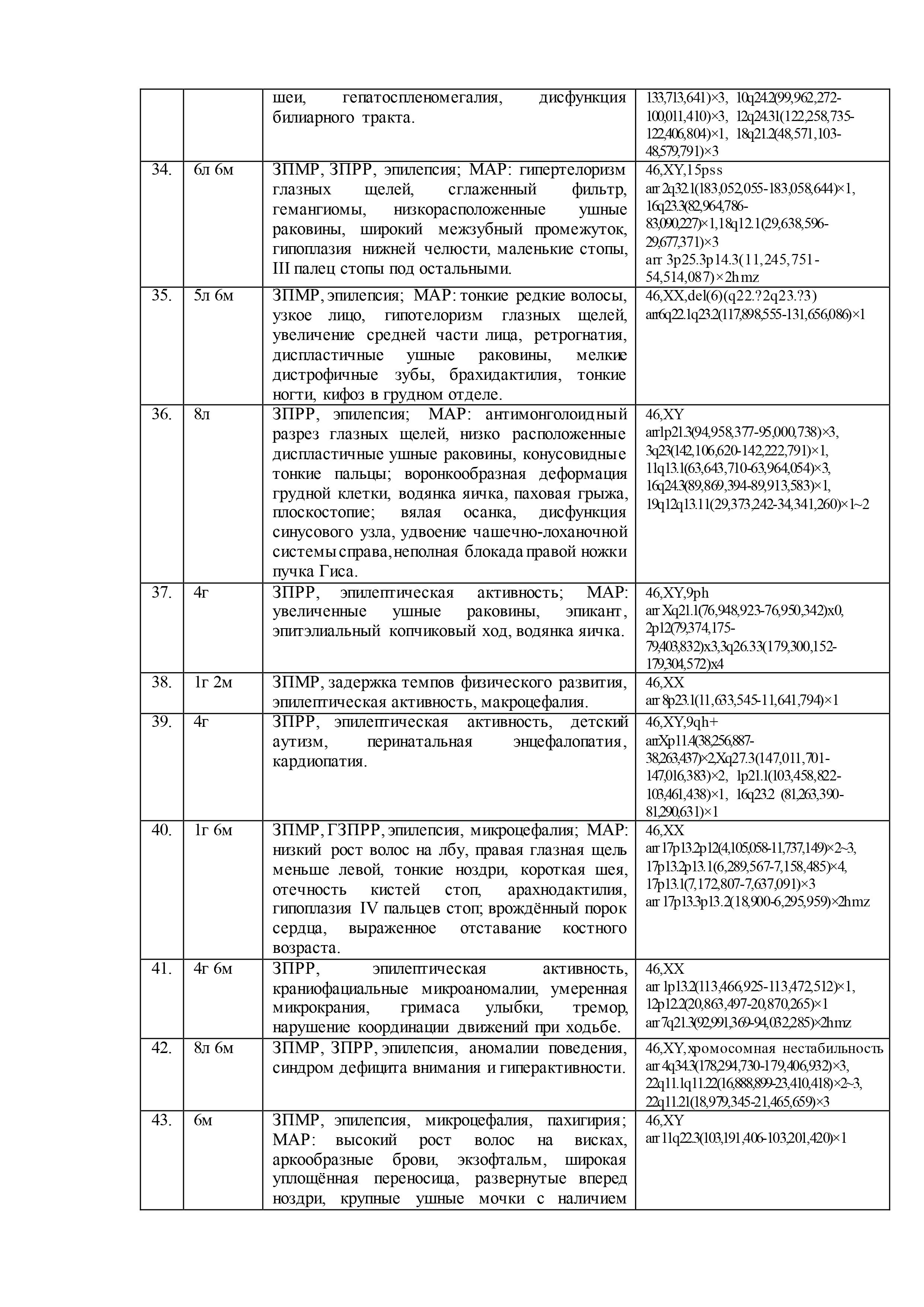
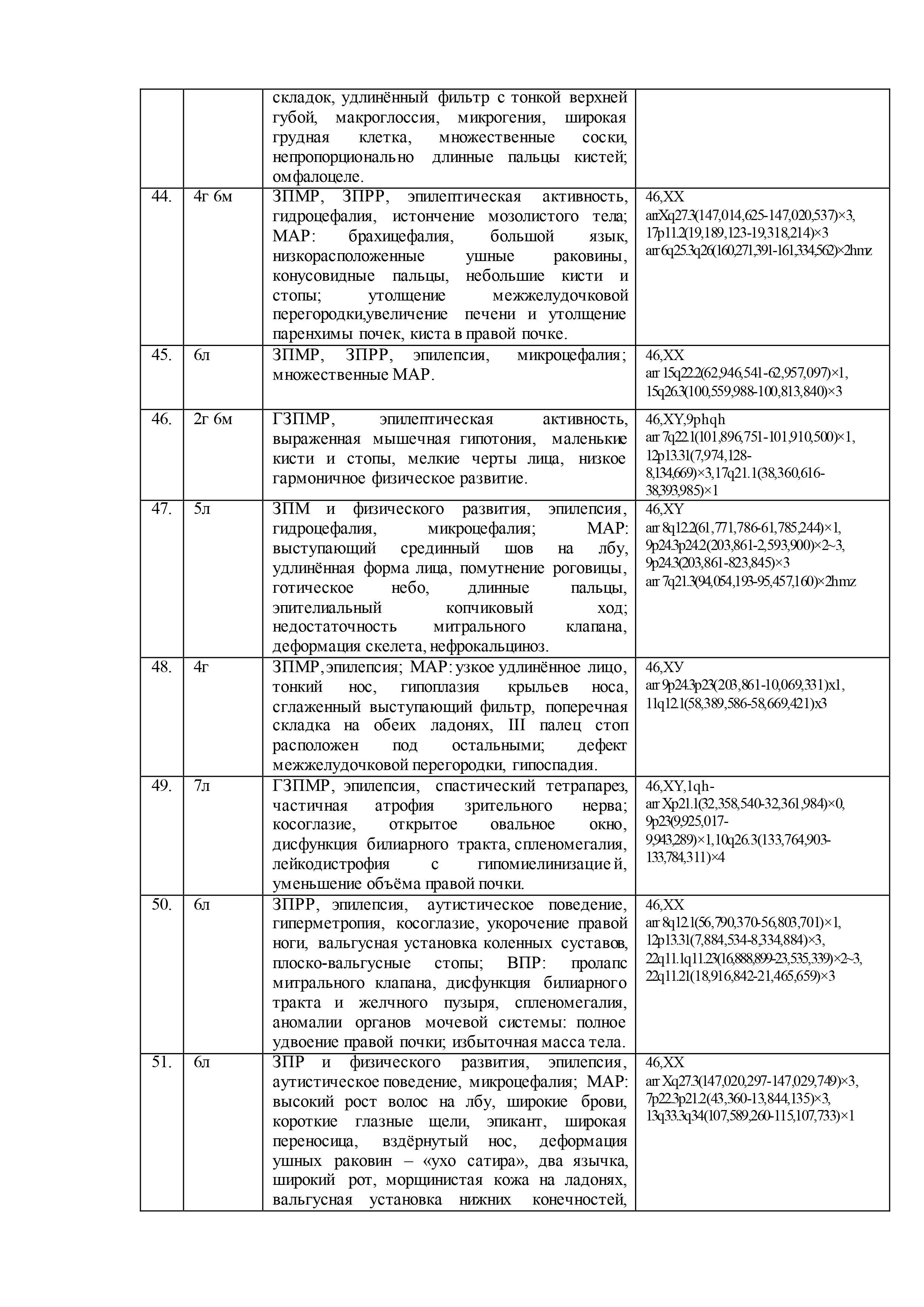
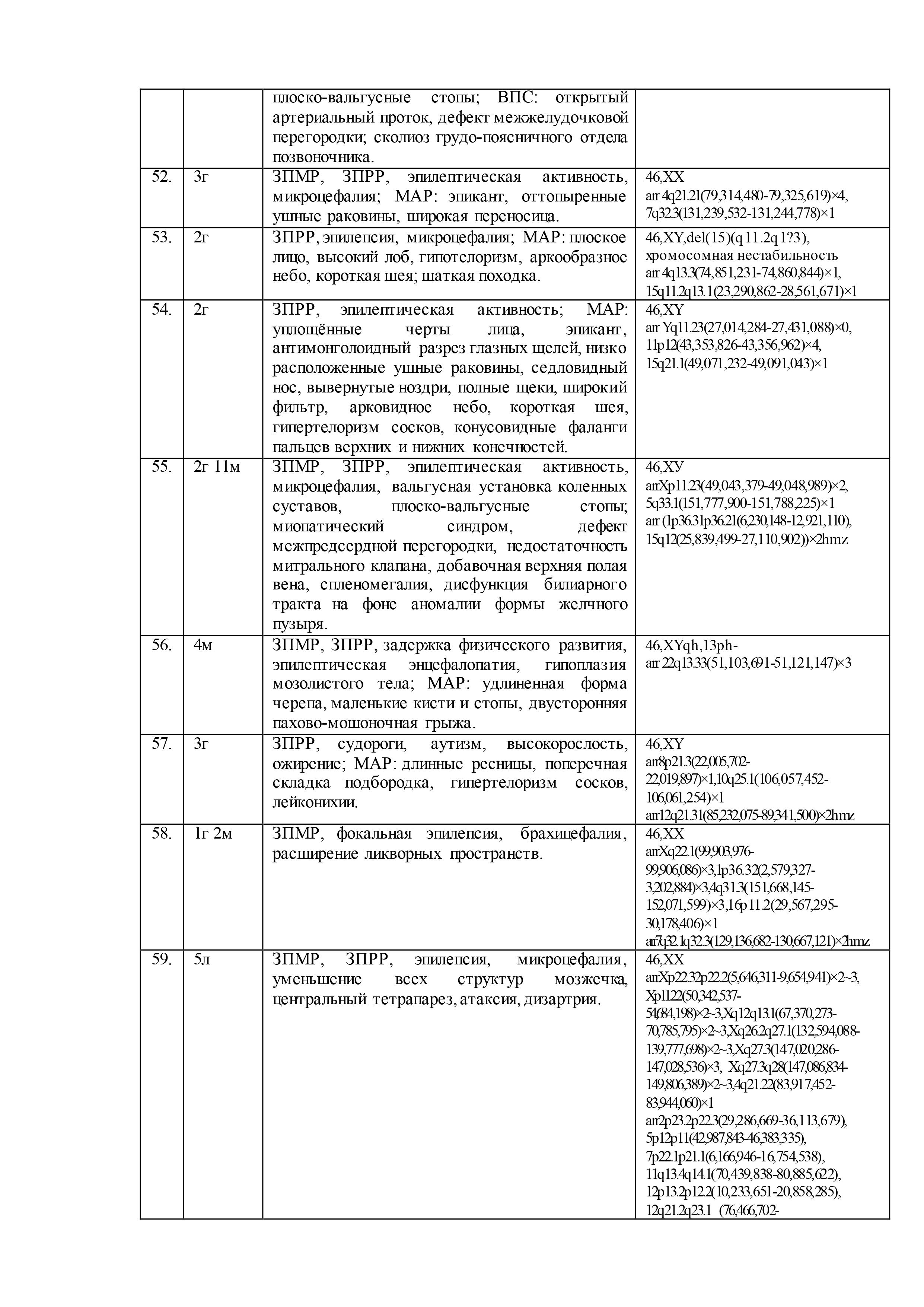

При цитогенетическом исследовании 61 ребенка c недифференцированными формами умственной отсталости и эпилепсией численные и структурные аномалии хромосом были выявлены у 5-ти (8%) пациентов: «дополнительная» маркерная хромосома –47,XX,+mar,13pss (случай №10 в таблице 2); «дополнительный» материал на хромосоме 3 –кариотип 46,XX,add(3)(p26),1qh+,21ps+ (случай №26); перестроенная хромосома 11 в виде инсерции с инверсией предположительно «дополнительного» материала –46,XX,der(11)?add(11)(p13)ins(11)(p13q21q23.3) (случай №30); делеция длинного плеча хромосомы 6 – 46,XX,del(6)(q22.?2q23.?3) (случай №35); делеция длинного плеча хромосомы 15 – 46,XY,del(15)(q11.2q1?3) (случай №53). Кроме того, у 7 пациентов (11%) была выявлена хромосомная нестабильность (№2, №4, №8, №12, №42, №53, №60), а в 21 случае (34%) – хромосомные варианты (изменения в гетерохроматиновых районах хромосом)(№3, №4, №8, №10, №11, №15, №18-20, №23, №26, №27, №32-34, №37, №39, №46, №49, №56, №60). Причем, у одного ребёнка хромосомная аномалия сочеталась с хромосомной нестабильностью (№53) и у двух– с хромосомными вариантами (№10, №26). У 3х детей хромосомная нестабильность была в сочетании с хромосомными вариантами (№4, №8, №60). В остальных случаях (56%) был выявлен нормальный кариотип.
После проведения молекулярного кариотипирования у 61 ребёнка было выявлено 192 аномалии генома с патогенным или вероятно патогенным эффектом на основании сравнения баз данных DGV, BioGPS, GenAtlas, KEGG и собственной базы данных лаборатории: у 20 детей (33%) – крупные аномалии (от 500 тысяч пн и более), у 29 детей (47%) – вариации числа копий последовательности ДНК (CNV), у 12 – интрагенные перестройки (20%). Кроме того, выявлено 23 участка сегментной потери гетерозиготности (унипарентальная дисомия) у 15 (25%) детей. При этом сочетанные аномалии генома наблюдались у 53 (87%) детей, а у 8 (13%) детей было выявлено по 1 аномалии генома. Среди CNV были обнаружены дупликации, делеции, мозаичные дупликации и делеции, трипликации.
Наиболее часто встречались аномалии генома, затрагивающие хромосому Х. Так, обнаружено 30 из 192 (15,6%) геномных аномалий хромосомы Xс патогенным или вероятно патогенным эффектом у 17 детей (28%) из 61 ребенка с недифференцированной формой умственной отсталости и эпилепсией. Среди геномных аномалий выявлены делеции, регулярные и мозаичные дупликации, трипликации. По хромосоме Y обнаружено 6 (3,1%) делеций и дупликаций у 5 детей (8%).
По хромосомам 1 и 2 обнаружено по 8 (4,2%) геномных аномалий в каждой из этих хромосому 8 детей (13%). Среди них были делеции, дупликации итрипликации. Аномалии хромосомы 3 встречались у 10 детей (16%), включая делеции, дупликации и трипликации, выявлено 10 (5,2%) геномных аномалий.
В хромосомах 4 и 5 обнаружено по 5 (2,6%) геномных аномалий в каждой из этих хромосому 5 детей (8%), среди них делеции, дупликации и трипликация по хромосоме 4, а по хромосоме 5 только делеции и дупликации.
По хромосоме 6 – 4 (2,1%) аномалий были обнаружены у 4 детей (6%), среди них три делеции и одна дупликация; по хромосоме 7 – 10 (5,2%) геномных аномалий у 10 детей (16%), среди них делеции, дупликации и трипликация; по хромосоме 8 – 10 (5,2%) аномалий у 8 (13%) детей, среди них делеции, дупликации и трипликация; по хромосоме 9 – 10 (5,2%) геномных аномалий у 7 (11%) детей, среди них делеции, регулярные и мозаичные дупликации и трипликации; по хромосоме 10 – 9 (4,7%) аномалий у 8 (13%) детей, среди них одна делеция, дупликации и трипликации; по хромосоме 11 – 11 (5,7%) геномных аномалий у 10 (16%) детей, среди них делеции, дупликации и трипликация; по хромосоме 12 – 8 (4,2%) аномалий у 8 (13%) детей, среди них делеции и дупликации.
По хромосоме 13 обнаружены 4 (2,1%) аномалии генома у 4 (7%) детей, среди них три делеции и одна трипликация; по хромосоме 14 наблюдались две делеции (1%) у двух детей (3%); по хромосоме 15 выявлено 10 (5,2%) геномных аномалий у 9 (14%) детей, среди них делеции и дупликации.
По хромосоме 16 обнаружено 10 (5,2%) аномалий генома у 8 (13%) детей, среди них делеции и дупликации; по хромосоме 17 – 15 (8%) аномалий генома у 12 (20%) детей, среди них делеции, регулярные и мозаичные дупликации, трипликация; по хромосоме 18 выявлены 2 (1%) дупликации у 2 (3%) детей.
По хромосоме 19 выявлено 9 (4,7%) аномалий у 9 (15%) детей, среди них делеции и дупликации. По хромосомам 20 и 21 геномных аномалий обнаружено не было. По хромосоме 22 наблюдалось 6 (3%) геномных аномалий у 4 (7%)детей, среди них регулярные и мозаичные дупликации.
Анализируя геномные аномалии, следует отметить, что наиболее часто они обнаружены в хромосоме X – в 15,6% и аутосомах 17 и 11, в 7,8% и 5,7%, соответственно. Меньше всего геномных аномалий выявлено по хромосомам 14 и 18 (по 1%). Как уже говорилось выше, по хромосомам 20 и 21 геномных аномалий обнаружено не было.
Следует отметить, что в представленной группе из 61 ребенка с недифференцированной формой умственной отсталости и эпилепсией среди 192 геномных аномалий было выявлено 97 делеций, что составило 50,5%, 71 дупликация (37%), 15 трипликаций (7,8%) и 9 (4,7%) мозаичных геномных нарушений, как правило, совместно с регулярными перестройками. В группе исследуемых детей мозаицизм выявлен по хромосомам 9,17,22 и Х. Чаще всего мозаицизм обнаружен в хромосоме Х (5 из 9 случаев), по хромосоме 22 – в двух случаях. Следует отметить, что с разработкой современных геномных технологий проблема мозаицизма интенсивно развивается многими лабораториями мира [14, 15, 16].
Примечательно, что в 9 (15%) случаях у детей были выявлены хромосомные (микроделеционные и микродупликационные) синдромы (рис.2).

Выявленные микроделеционные и микродупликационные синдромы не были идентифицированы при клинико-генетическом обследовании. Возможно, это связано с наличием других различных сочетанных аномалий генома (CNVs), а не только с нарушениями в критических локусах, ассоциированных с конкретным синдромом (№10, №13, №20, №30, №42, №44, №50, №53, №61). Следует отметить, что даже при наиболее часто встречающихся микроделеционных/микродупликационных синдромах, затрагивающих соответствующие области генома, вклад отдельных генов с другой локализацией в формирование патологических фенотипических проявлений изучается крайне редко, и может быть различен в связи с определёнными повреждениями других участков генома. Среди обнаруженных синдромов были микроделеции в участке 2q24.3, синдромы Ангельмана (15q11.2-q13), Кулена-Де Вриза (17q21.31) – по 2 случая, а также микродупликации – синдром Потоки-Лапски (17p11.2p13.3) и велокардиофациальный синдром (22q11.2) – по 2 случая. Причем, в одном случае наблюдалось сочетание синдрома микроделеции 2q24.3 с синдромом Ангельмана.
Известно, что использование биоинформатических методов является необходимым при интерпретации полученных с помощью молекулярного кариотипирования данных для определения молекулярных механизмов заболеваний. Одним из биоинформатических подходов к идентификации патогенности геномных перестроек и ассоциации вариабельности последовательности ДНК (CNV) с определенными фенотипическими признаками, является приоритизация генов-кандидатов [10-13]. Поскольку функциональные характеристики генов являются относительно постоянными параметрами, совокупность выявленных данных относительно изменений числа их копий может быть использована для оценки последствий генных и хромосомных (геномных) мутаций у детей с недифференцированными формами умственной отсталости, ВПР и/или МАР и эпилепсией (эпилептической активностью) и, возможно, в дальнейшем позволит осуществить прогнозирование и коррекцию клинических проявлений у ребёнка.
В исследуемой группе из 61 ребёнка при применении оригинального биоинформатического анализа с использованием электронных ресурсов DGV, BioGPS, GenAtlas, KEGG, OMIM и собственной базы данных лаборатории выявлено большое число генов (более 800), среди которых из повторяющихся генов были следующие: чаще всего наблюдался ген FMR1 [OMIM:309550], ассоциированный с умственной отсталостью, сцепленной с ломкой хромосомой X,при которой часто выявляют эпилепсию (эпилептическую активность); затем гены DAZ2[OMIM:400026], DAZ3[OMIM:400027] в участкеYq11.223Yq11.23 (4 делеции и 2 дупликации), при этомданные делеции затронули локус, расположенный в критическом участке хромосомы Y, потери которого ассоциируютcумственной отсталостью и аутизмом [7], делеции и дупликации в данном локусе также ассоциированы с мужским бесплодием (азооспермией и/или олигоспермией) [17, 18]. Обнаружен также ген BTRC, связанный с передачей сигналов (бета-трансдуцированный белок) [OMIM:603482], в участке 10q24.32.Этот ген вовлечен в 7 геномных сетей (базы данных KEGG), один из них – сигнальный путь циркадного ритма (circadian rhythm), который связан с эпилептическими проявлениями, поскольку ритмические паттерны в эпилептической активности и возникновении приступов ассоциированы с циркадными колебаниями в возбуждающем и тормозном равновесии. Так, было показано, что гены BMAL1 и CLOCK, входящие в циркадный сигнальный путь, влияют на порог возбудимости нейронов головного мозга и возникновения судорог [19].
Среди повторяющихся генов были гены, ранее связанные как с умственной отсталостью, так и эпилепсией: ген AFF2 (FMR2) [OMIM:300806] – в участке Xq28. Вариации числа копий последовательности ДНК данного гена ассоциированы с Х-сцепленной умственной отсталостью FRAXE [OMIM:309548]. Ген SLC1A1 [OMIM:133550] локализован в участке 9p24.2 и кодирует белок, который играет значимую роль в транспортировке глутамата через плазматические мембраны, а также ассоциирован с различными заболеваниями головного мозга, в том числе и с шизофренией [OMIM:615232]. Обнаружены также гены SCN2A [OMIM:182390] и SCN3A [OMIM:182391], локализованные в участке 2q24.3 и кодирующие один из белков натриевого канала; нарушения в данных генах ассоциированы с эпилептическими приступами [20]. Ген NECAP1 [OMIM:615833] был выявлен в хромосомном участке 12p13.31, нарушения в этом гене ведет к ранней инфантильной эпилептической энцефалопатии [21]; генGABRB3 [OMIM:137192], локализованный в участке 15q11.2q12 [OMIM:612269], ответcтвенен также за раннюю инфантильную эпилептическую энцефалопатию. Ген SHANK3 [OMIM:606230] локализован в участке 22q13.33: при этом, белок ассоциирован с рецепторами нейротрансмиттеров ионных каналов и с сигнальными путями, связанными с G-белком. Описываются случаи, когда мутации гена SHANK3 вызывают нарушения развития и эпилепсию [22]. Причем, часто гены локализованы в одних и тех же участках с геномными аномалиями и расположены относительно «близко» друг от друга, как например ген UBE3A [OMIM:601623], ассоциированный с синдромом Ангельмана, и ген GABRB3 [OMIM:137192], ассоциированный с ранней инфантильной эпилептической энцефалопатией, локализованы в участке 15q11.2q12 [OMIM:612269].
Анализируя полученные результаты, следует отметить, что часто повторяющиеся гены (FMR1, DAZ2и DAZ3), обнаруженные в нашем исследовании, связаны с такими клиническими проявлениями, как умственная отсталость с аутистическими расстройствами, ВПР и/или МАР. Гены DAZ2 и DAZ3 локализованы в хромосоме Y и связаны также с азооспермией и олигоспермией. Следует отметить, что ген BTRC связан непосредственно с эпилептической активностью.
Таким образом, исходя из полученных результатов, вариабельность полученных клинических и молекулярно-цитогенетических данных не позволяет пока провести корректные сопоставления патологической значимости геномных нарушений в ассоциации с недифференцированными формами умственной отсталости и эпилепсии. Тем не менее, подобные исследования следует продолжать с целью накопления материала по геномным перестройкам в каждом конкретном случае (персонифицированная геномика). Совместное использование методов, направленных на исследование индивидуальных и межклеточных вариаций генома в сочетании с оригинальным биоинформатическим анализом, определяет патогенетическое значение вариаций генома и основную причину многих фенотипических проявлений (таблица 2).
Заключение. Полученные нами данные обследования 61 ребенка c недифференцированными формами умственной отсталости и эпилепсией, ВПР и/или МАР выявили различные аномалии генома, в том числе вариации числа копий последовательности ДНК (CNVs), что позволило предположить роль этих геномных аномалий в клинических проявлениях (таблица2). При цитогенетическом исследовании обнаружены численные и структурные аномалии хромосом в 8% случаев.
При молекулярном кариотипировании у 61 ребенка выявлено 192 аномалии генома с патогенным или вероятно патогенным эффектом. При этом сочетанные аномалии генома наблюдались в 87% случаев и только у 13% детей было выявлено по одной аномалии генома. Среди 192 геномных аномалий было выявлено в 50,5% делеции, 37% дупликации, 7,8% трипликации и 4,7% мозаичные геномные нарушения, как правило, совместно с регулярными перестройками. Геномные аномалии, обнаруженные в процессе исследования, встречались по всем хромосомам, кроме хромосом 20 и 21.
При применении оригинального биоинформатического анализа, используя приоритизацию генов, определено более 800 генов, среди них из повторяющихся генов были следующие: ген FMR1 [OMIM:309550], ассоциированный с умственной отсталостью, сцепленной с ломкой хромосомой X; затем гены DAZ2 [OMIM:400026] и DAZ3 [OMIM:400027]. Особо следует отметить выявленный ген BTRC [OMIM:603482], непосредственно связанный с эпилепсией. Реже выявлены гены AFF2 (FMR2) [OMIM:300806], SLC1A1 [OMIM:133550], SCN2A [OMIM:182390], SCN3A [OMIM:182391], GABRB3 [OMIM:137192], NECAP1 [OMIM:615833], SHANK3 [OMIM:606230]. Вариабельность полученных данных и число обследованных детей не позволяют провести корректные корреляции патологической значимости геномных нарушений в плане недифференцированных форм умственной отсталости и эпилепсии. Однако результаты работы показывают, что подобные исследования следуют проводить с целью накопления результатов по полногеномному анализу при умственной отсталости и эпилепсии. Более того, предыдущие исследования генетических синдромов с различными формами эпилепсии (например, синдром Ретта), а также умственной отсталости и аутистических расстройств в сочетании с эпилептиформными проявлениями показали, что поиск молекулярных механизмов соответствующей патологии головного мозга имеет наибольший смысл именно в «полногеномном» контексте [23, 24, 25].
Накопление данных по генетическим исследованиям – совместное использование цитогенетических, молекулярно-цитогенетических и углублённых биоинформатических технологий для идентификации индивидуальных и межклеточных вариаций генома с анализом клинических проявлений – позволит определить механизм заболевания в каждом отдельном случае (персонифицированная геномика) и выявить возможные корреляции у детей с идиопатическими формами умственной отсталости и эпилепсией.
Следует отметить, что биоинформатические методы необходимы для интерпретации данных, полученных с помощью молекулярного кариотипирования, одним из подходов которого к идентификации патогенности геномных перестроек является приоритизация генов. Данные относительно изменений числа копий последовательности ДНК в дальнейшем можно использовать для оценки фенотипических последствий генных и хромосомных (геномных) мутаций у детей с идиопатическими формами умственной отсталости и эпилепсии. Полученные нами результаты и проведённый анализ может указывать на целесообразность и продолжение исследований, направленных на изучение недифференцированной умственной отсталости и эпилепсии.














Список литературы