
2q14 de novo interstitial deletion in a girl with intellectual disabilities and malformations
Aннотация
Background: Neurodevelopmental disorders have a prevalence of approximately 3% in the population, the genetic factor is involved in this condition. Microdeletions that comprise the 2q14.1q14.3 region are rare events. The aim of the study: To describe a girl with an interstitial deletion of the 2q14 region with a severely affected phenotype and to compare her with reports from the international literature. Materials and methods: Cell culture and the obtaining of chromosomes were carried out according to the standardized techniques in our laboratory. Results: Girl evaluated for dismorphias and brain malformation (corpus callosum agenesis). The chromosomal study with a resolution of 565 bands showed an interstitial deletion in the long arm of chromosome 2. Karyotype: 46, XX, del (2) (q14.1q14.2). Conclusion: The correct clinical analysis of the patient and the high-resolution cytogenetic technique has been successful for detection of the microdeletion in this case helping in genetic counseling and in elucidating the genetic origin of the patient's neurodevelopmental disorder.
К сожалению, текст статьи доступен только на Английском
Introduction. Neurodevelopmental disorders (NDD) have an estimated prevalence of about 3% in worldwide population and they are considered a health problem in countries with a well-implemented public health service. This condition has a high incidence of the genetic factor, since about 28% of patients with NDD present some type of abnormality in their genome [1]. Until present date, up to 450 different genes have been described and found t to be directly involved in NDD, and it is estimated that many more are to be discovered [2]. Numerous microdeletion-microduplication syndromes (MMSs) involving new genes have been identified using molecular methods and a high-resolution technique on chromosomes. For most NDD cases, intellectual disability has been found to be the most common feature [3].
Microdeletions that comprise the 2q14.1q14.3 chromosomal region are rare events. Until present date, the reviewed literature only reports nine similar cases, some with large deletions of more than 20 Mb, and others with cryptic deletions smaller than 6 Mb that are not detected by conventional cytogenetics. Depending on the extension and location of the lost segment in this region, the phenotype of the patients may be subject to variability. However, there are characteristics common to most affected individuals, such as: intellectual disabilities of different degrees, postnatal growth retardation and facial dysmorphism [4].
The aim of the study. In the present study we describe a case with an interstitial deletion of the 2q14 region, de novo, in a girl with a severely affected phenotype and compare it with reports found in international literature.
Materials and methods. Cell culture and the obtaining of chromosomes were carried out according to the standardized techniques in our laboratory. Chromosome analysis is performed with a resolution greater than 550 bands.
Results and discussion. The girl evaluated for dysmorphia and brain malformation was born by caesarean section due to pelvic cephalous disproportion at 39 weeks’ gestation.
First daughter of non-consanguineous couple, there is no pathological history in the family. Follow-up during pregnancy with normal alpha fetoprotein analysis and ultrasound during the third trimester present an apparent macrocephaly and dilatation of the ventricular system.
At birth, height and weight were adequate for gestational age. During postnatal follow-up, hypotonia and poor sucking reflex were noted. The girl was admitted to neonatology for omphalitis. The girl was examined by specialists who diagnosed the symptoms of sustained hypotonia.
Physical examination at four months of age showed facial dysmorphias such as hypertelorism, a depressed nasal bridge, anteverted nostrils, a wide anterior fontanel and an open posterior fontanelle. Normal hematological parameters.
Transfontanelle ultrasound: a dilatation of the ventricular system and absence of the corpus callosum were detected.
Skull IMR: cerebral malformation with poor differentiation between white and gray matter, hydrocephalus and absence of the corpus callosum.
Karyotype
By means of a chromosomal study with a resolution of 565 bands, an interstitial deletion in the long arm of chromosome 2 (46, XX, del (2) (q14.1q14.2) was diagnosed. Band 2q14.3 is conserved in this patient (Figure).

The parents have a normal phenotype, the chromosomal studies of both were normal.
Genotype - Phenotype Correlation
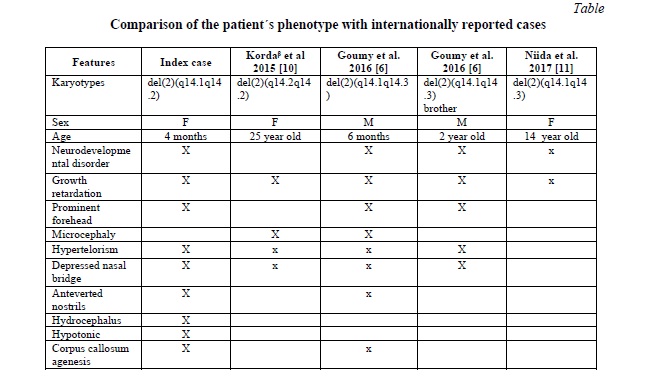
The patients in Table 1 share phenotypes that indicate malformations in the structural organization of the nervous system. A spectrum from large malformations that include the ventricular system to defects that cause the absence of the corpus callosum is observed in all patients. Furthermore, a series of common phenotypic characteristics are present, such as: growth retardation, prominent forehead, hypertelorism, depressed nasal bridge and intellectual disability.
A girl with severe neurodevelopmental disorders and other large malformations, such as corpus callosum agenesis, is described. Additionally, she presents several dysmorphic features and an unusual interstitial deletion in 2q14 which was diagnosed by high-resolution methods. A comparison with similar cases reported in the literature has been made to define affected genes which are responsible for the phenotype.
Gomumy et al. (2016) report the case of a boy and his brother with a deletion of the 2q14.1q14.3 segment diagnosed by microarray studies. According to these authors, the deleted area comprises 5.8 Mb of the genome and contains 79 genes, of which 24 are coding and three genes are described in the OMIM as morbid genes, due to their haploinsufficiency: STEAP3, GLI2, and RNU4ATAC [4].
The STEAP3 gene has a role as a metal reducer and participates in the homeostasis of iron present in erythrocytes. The deficiency of this gene causes microcytic-hypochromic anemia [5]. The two patients reported by Gomumy and the one described in present study had no clinical signs of hypochromic anemia. This suggests that this gene is not found within the deleted genes in our patient.
The GLI2 gene encodes a protein which, when deficient, for instance due to a mutation has been associated with characteristics of the spectrum of holoprosencephaly and has great phenotypic variability (including abnormalities in the formation of the pituitary gland and craniofacial abnormalities) [6]. Also in these patients, agenesis of the corpus callosum has been reported [7]. The patient reported in this study presents marked hypertelorism, which does not correspond to the holoprosencephaly referred in these cases. It is very likely that this gene is not deleted in the patient described here.
The RNU4ATAC gene encodes a small nuclear RNA involved in the activity of correct splice and separation of introns to form messenger RNA. Mutations of this gene lead to intron splicing errors of approximately 800 types of U12 introns involved in a wide variety of cellular processes [8]. These alterations cause Taibi-Linder syndrome characterized by pre and post-natal growth retardation, microcephaly, severe brain malformations, dysmorphic features, bone dysplasia and hearing and vision defects. Due to this mutation in the RNU4ATAC gene, all patients have partial or complete absence of the corpus callosum [9] This major malformation is present in our patient and coincidentally is associated with the haploinsufficiency of the gene previously described. In addition, other phenotypic characteristics of the case in question coincide with those of patients who carry mutations in the RNU4ATAC gene (Table).
Corpus callosum agenesis has been described as a consequence of large deletions including the 2q14 region. Antich et al. [12] describes the deletion from 2q12-q14, Frydman et al. [13] from 2q14-q21 and Greally et al. [14] from 2q14-q22.1. The latter has found this malformation in two of the 9 patients he reports. Another characteristic present in patients with deletion in the 2q14 region is the growth retardation that has also been found in patient under study. Greally et al suggested that it was due to haploinsufficiency of the GLI2 gene [14].
Kordaᵝ et al. [10] describe a family with the small deletion of 4.3 Mb in the 2q14.2 region that includes the GLI2 and RNU4ATAC genes. Several dysmorphic features are described but none belonging to the spectrum of holoprosencephaly. The authors confirm that it is due to the incomplete penetrance that characterizes the GLI2 gene mutations [6, 15]. The family described by Korda et al. does not present intellectual disability and in the index case the most striking was the postnatal growth retardation, a feature that is also present in this girl.
Even when the limitations of the method used to make the diagnosis in the present case are recognized, the karyotype-phenotype correlation and its coincidence with the reports in the scientific literature suggest that the detected deletion in the 2q14 region is a certain possibility for a correct diagnosis of this case. Agenesis of the corpus callosum does not occur in all the patients reported in the consulted literature, perhaps due to the phenomenon of incomplete penetration of The GLI2 gene or perhaps because the RNU4ATAC gene is not affected by the deletion, which is a possibility still to be defined.
Since this major malformation is present in this patient, confirmation of the diagnosis by more precise molecular methods is recommended in order to classify this case as example of a 2q14 deletion and to define a possible MMS with this designation in the future.
Conclusions. The correct clinical analysis of the patient and the high-resolution cytogenetic technique has been successful for detection of the microdeletion in this case helping in genetic counseling and in elucidating the genetic origin of the patient's neurodevelopmental disorder.
Благодарности
The authors want to recognize the support of the RFBR and CITMA institutions of Russia and Cuba respectively for our study














Список литературы
Список использованной литературы появится позже.