
Синдром делеции короткого плеча хромосомы 18 (18р-) у детей: возможности цитогенетической и молекулярно-цитогенетической диагностики
Aннотация
Актуальность: Синдром делеции короткого плеча хромосомы 18 (18р-) связан с различными потерями хромосомного материала короткого плеча (частичная моносомия), но чаще всего с полной потерей короткого плеча хромосомы 18. Частота синдрома 18р- в популяции составляет 1:60000; цитогенетически и клинически он достаточно гетерогенен. Клинические проявления значительно вариабельны: от лёгких форм с врождёнными пороками и микроаномалиями развития до грубых пороков головного мозга; редко наблюдаются расстройства аутистического спектра, эпилепсия. Точки разрыва при делеции разнообразны, так что синдром требует корректного исследования больших групп больных детей с применением современных геномных технологий. Цель исследования: Использование цитогенетических и молекулярно-цитогенетических технологий для определения критических точек разрыва и, по возможности, корреляции фенотипа и генотипа при синдроме 18р-. Результаты: В данной публикации мы описываем собственные наблюдения 15-ти пациентов (9 мальчиков и 6 девочек) с синдромом делеции короткого плеча хромосомы 18, выявленных в обширной когорте обследованных пациентов (n=8536). Средний возраст детей составил 5,1г; соотношение полов был в пользу мальчиков (1,5:1) в отличие от литературных данных. Критических точек, связанных с этим синдромом и локализованных в коротком плече хромосомы 18, не выявлено. Вероятно, клиническая картина синдрома ассоциирована со многими точками разрыва в коротком плече 18(р11.1->pter). Частота синдрома 18р- в исследуемой группе детей с задержкой развития, и умственной отсталостью, врождёнными пороками и микроаномалиями развития составила 0,2%. Обсуждаются диагностические аспекты данной патологии и возможности применения молекулярно-цитогенетических методов в исследовании синдрома. Заключение: Подчёркивается персонализированный подход к диагностике синдрома для корректного медико-генетического консультирования с целью улучшения качества жизни больного ребёнка и для дальнейшего изучения корреляций фенотип-кариотип
Ключевые слова: синдром 18р-, цитогенетические методы, молекулярно-цитогенетическая диагностика, умственная отсталость, врождённые пороки и аномалии развития
Введение. В 1960-х годах прошлого века были описаны синдромы, связанные с аномалиями хромосомы 18, в том числе синдромы 18p- и 18q-, а также синдром кольцевой хромосомы 18 (r18) [1, 2]. Основные патологические проявления синдрома делеции короткого плеча хромосомы 18 (18р-) связаны с различными психомоторными и другими врождёнными пороками и микроаномалиями развития. Учёные из разных стран, исследовавшие большое число детей с данной аномалией, отмечают значительный цитогенетический и клинический полиморфизм, частые стёртые формы синдрома, нерешённые проблемы корреляции фенотип-генотип, что затрудняет медико-генетическое консультирование, коррекционную терапию, прогноз для пациентов, и требует применения самых современных цитогенетических и молекулярно-цитогенетических методов для более эффективного изучения данного синдрома [2].
Минимальными диагностическими признаками этого синдрома считают умственную отсталость от лёгкой до тяжёлой степени, задержку роста, мышечную гипотонию, крупные деформированные ушные раковины, аномалии скелета. Выделяют два основных фенотипических варианта данного синдрома: 1) с грубыми пороками аринэнцефалической системы; 2) с отсутствием пороков мозга. Типичны также следующие признаки: низкая масса тела при рождении, задержка роста, укороченные конечности, различные МАР: микроцефалия, гипертелоризм глазных щелей, птоз, эпикант, катаракта, косоглазие, широкий плоский нос, микрогнатия, расщелины губы и нёба, большие оттопыренные ушные раковины, нарушения строения челюстно-зубного аппарата, множественный кариес, клинодактилия, синдактилия; нередки короткая шея, широкая вдавленная грудная клетка; алопеция, гипопигментация кожи, аномалии позвоночника, вальгусная деформация локтевых суставов, косолапость, врождённый вывих бедра, гипоплазия мошонки и полового члена у мальчиков и малых половых губ у девочек, паховая грыжа, ВПС. Все больные значительно отстают в психомоторном развитии, страдает речь (афазия или дисфазия, фразовая речь часто отсутствует до 7-9 лет). Встречаются грубые пороки головного мозга при первом варианте заболевания (аринэнцэфалия, прозэнцефалические пороки, гипоплазия гипофиза - гипопитуитаризм, циклопия, цебоцефалия) [3, 4, 5]. У этих пациентов снижена продолжительность жизни. Частота синдрома составляет 1:60000, девочки болеют чаще мальчиков (соотношение полов по разным данным: М:Ж – 1:1,5 - 1:2) [6, 7, 8]. Большинство случаев описаны как спорадические; семейные случаи редки и обычно ассоциированы со сбалансированными перестройками родителей при участии 18р. Редко несбалансированная перестройка наследуется от одного из родителей со сбалансированной аномалией, связанной с 18р, и тогда родители могут иметь подобные клинические симптомы синдрома в лёгкой форме [9]. Прогноз для жизни у больных с пороками головного мозга неблагоприятен, возможна гибель таких пациентов в первые месяцы и даже дни жизни, у больных без пороков мозга продолжительность жизни нормальна или снижена незначительно (известны случаи смерти в возрасте 65 лет) [2]. Отдельные больные хорошо адаптируются и даже способны к деторождению. Родители больных детей в среднем несколько старше обычного и их возраст составляет на момент рождения ребёнка более 35-36 лет. Поскольку у многих пациентов с делецией короткого плеча хромосомы 18 клинические признаки весьма вариабельны, диагностика синдрома нередко затруднительна и требует подтверждения молекулярно-цитогенетическими методами [4, 10, 11].
Подробное изучение генов в коротком плече хромосомы 18 позволяет анализировать их участие в геномных сетях, которое, в свою очередь, может быть предметом обсуждения при поиске корреляций генотип-фенотип при этом синдроме. По данным исследований [4], в коротком плече хромосомы 18 содержится 118 генов, 57 из которых индексированы в базе данных OMIM [Online Mendelian Inheritance in Man]. Большинство из них не участвуют в формировании патологического фенотипа при данном синдроме. Авторам удалось идентифицировать 10 генов, которые, с большой вероятностью, участвуют в формировании фенотипа при данном синдроме: TGIF1, LAMA1, GNAL, AFG3L2, SMCHD1, PTPN2, TWSG1, DLGAP1, ANKRD12 и IMPA2. Тем не менее, многие исследователи полагают, что у данного синдрома нет критического участка или же критический участок расположен от теломеры до точки разрыва в каждом конкретном случае. Для отдельно взятых патологических симптомов (тугоухость, страбизм, птоз, нистагм, сколиоз/кифоз, крипторхизм, судороги) такие сегменты хромосомы 18 определены [7, 12, 13]. Нередко детям с синдромом 18р- клинически ставят диагноз синдром Шерешевского-Тернера, имея ввиду низкий рост, короткую шею, аномалии гениталий [14, 15, 16]. Цитогенетически синдром достаточно гетерогенен. Обычно делеция происходит с потерей всего короткого плеча, размер которого составляет 16000000-16500000 пар нуклеотидов, включая гетерохроматин центромеры. Реже разрывы при делеции наблюдаются в различных сегментах участка 18р11.21->pter. Известно также, что не выявлено ни одного случая интерстициальных делеций в коротком плече хромосомы 18.
В настоящей работе представлены цитогенетические и молекулярно-цитогенетические исследования детей с различными формами синдрома 18р-.
Материалы и методы исследования. Цитогенетически обследованы 8536 детей с ЗПРР, ЗПМР, умственной отсталостью, ЗФР, ВПР и МАР, среди которых мальчиков - 4575, девочек – 3961. Соотношение полов М:Ж – 1,15:1. Кариотипирование было проведено всем пациентам путём GTG- и CBG-окрашивания по стандартным протоколам [2]. Запись кариотипов проводилась согласно классификации и номенклатуры ISCN 2016 и 2020 [17, 18].
Молекулярно-цитогенетические исследования (FISH) проводились с помощью оригинальных методов и с применением оригинальных сайт-специфичных и околоцентромерных ДНК проб, а также в отдельных случаях - пэйтинговой пробы, по стандартным протоколам [19, 20].
Молекулярное кариотипирование проводили с помощью SNParray (CytoScan HD, Affymetrix), содержащей около 2,7 млн проб, по стандартным протоколам. Полученные данные визуализировались с использованием Affymetrix ChAS (Chromosome Analysis Suite, Array Version 4.1.0.90/r29400) [21, 22].
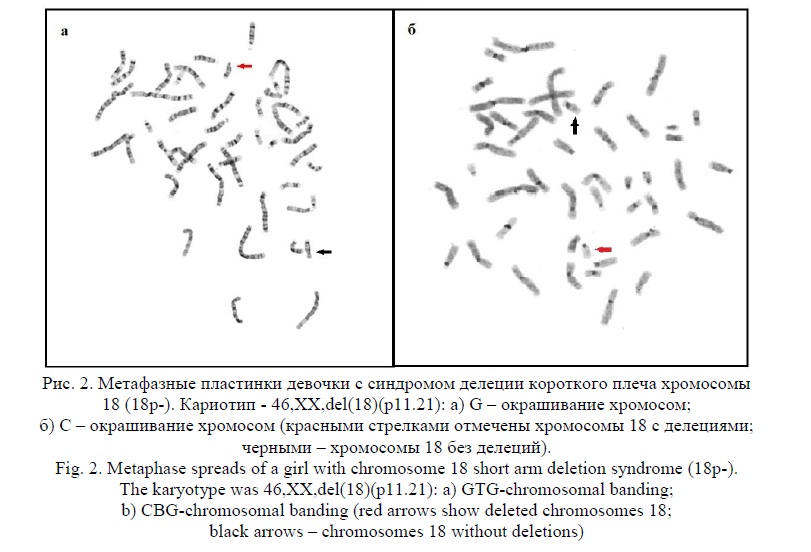
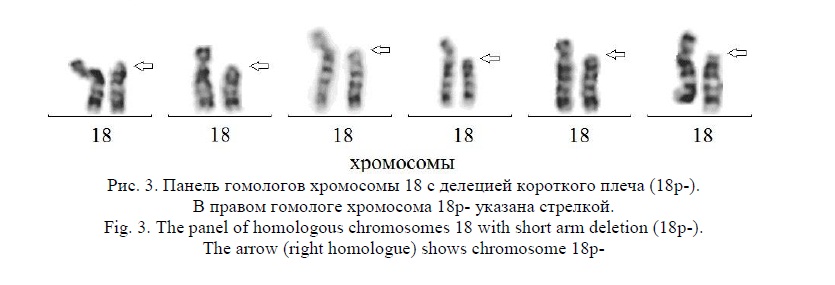
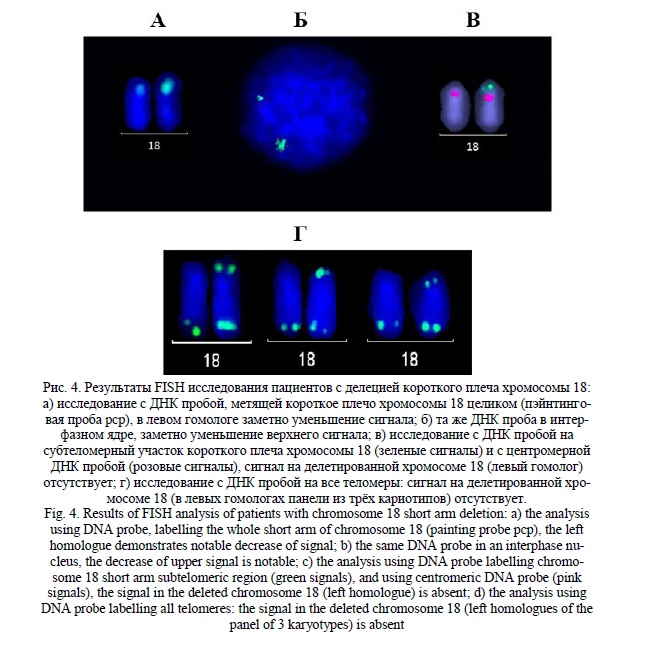
Результаты и их обсуждение. В результате цитогенетических исследований 8536 детей с ЗПМР (умственной отсталостью), различными ВПР и МАР было выявлено 15 детей (9 мальчиков и 6 девочек) с синдромом короткого плеча хромосомы 18. Соотношение полов - 1,5:1,0 было в пользу мальчиков в отличие от литературных данных, где всегда преобладают девочки. Средний возраст детей с синдромом делеции короткого плеча хромосомы 18 (18p-) составил 5 лет 1 месяц (от 3 месяцев до 12 лет). Частота детей с синдромом делеции короткого плеча хромосомы 18 (синдром 18р-) составила 0,2% от общего числа пациентов (15 пациентов из 8536 детей). При цитогенетическом анализе делеция (потеря хромосомного материала короткого плеча хромосомы 18) выявлена, как с точками разрыва, локализованными в районе 18р11.1 с участием околоцентромерного гетерохроматина, так и в участках 18р11.2 – 18р11.3 до 18pter (табл. 1). У 11-ти детей обнаружена делеция короткого плеча хромосомы 18, у 2х – кольцевая хромосома 18 с потерей хромосомного материала короткого плеча, у одного ребёнка – транслокация при участии хромосомы 18 с потерей части материала короткого плеча. Транслокация и кольцевые хромосомы (табл.1, случаи 3, 10 и 11) подтверждены и уточнены с помощью FISH исследований при применении сайт-специфических ДНК проб. В 5-ти из 15-ти случаев было утрачено всё короткое плечо хромосомы 18 (табл. 1). В одном случае выявлена мозаичная форма синдрома. Клинические симптомы у всех пациентов были разнообразны: умственная отсталость, ЗПРР, ЗПМР, ЗФР, РАС, анэнцефалия, различные ВПР и МАР, в том числе микроцефалия, низкий рост, глазокожный альбинизм, алопеция, гипертелоризм глазных щелей, расщелины губы и нёба и другие различные МАР (табл. 1). Степень тяжести патологических проявлений, включая пороки мозга и судороги, значительно варьировала; при этом, не все перечисленные аномалии (кроме ЗПМР и ЗПРР) встречались у больных детей [12, 23, 24]. Корреляция между размерами утраченного участка и тяжестью клинических проявлений отчётливо не прослеживалась, что объяснимо некоторой стёртостью клинической картины и разнообразием патологических симптомов у пациентов из данной группы (табл. 1). Практически, во всех случаях проводили FISH исследования, а в отдельном случае и молекулярное кариотипирование, и с помощью этих методов делеция короткого плеча хромосомы 18 была подтверждена. По цитогенетическим и FISH исследованиям из 15ти случаев обнаружены, как регулярные хромосомные аномалии, у 14ти пациентов, так и у одного ребёнка - мозаичная форма синдрома с клонами 46,ХУ,18р-/47,ХУ,+mar(derX) (последний клон обнаружен в небольшом количестве). Кроме того, у одного ребёнка выявлена транслокация с участием хромосом 15 и 18 с потерей короткого плеча хромосомы 18, и два случая с кольцевой хромосомой 18 также с потерей короткого плеча. При молекулярном кариотипировании обнаружена и частичная дупликация хромосомы 7 (табл. 1, случай 14). Результаты цитогенетических исследований представлены на рисунках 1, 2, 3. Причём, на рисунке 3 представлена панель гомологов хромосомы 18 из разных кариотипов, один из гомологов которых с 18р- указан стрелкой. FISH исследования представлены на рисунке 4.

В одном случае (табл.1) пациенту с 18р- было вместо FISH исследования проведено молекулярное кариотипирование (SNParray), которое не только подтвердило делецию короткого плеча хромосомы 18, но и позволило выявить другие геномные аномалии, в том числе дупликацию хромосомы 7 и CNV, которые, возможно, внесли свой вклад в клиническую картину синдрома.


FISH исследования представлены в таблице и на рисунке 4. В качестве примера диагностики методом молекулярного кариотипирования можно привести 14й случай из таблицы у мальчика 8ми лет, которому были проведены цитогенетическое и молекулярно-цитогенетическое исследования. Клинические признаки у ребёнка были следующие: ЗПРР, СДВГ, черты аутизма, левосторонний нефроптоз, функциональные нарушения ЖКТ, синусовая тахикардия, диффузный зоб, аномалия шейного отдела позвоночника. Для поиска геномных аномалий и для уточнения точек разрыва при делеции 18р пробанду провели молекулярное кариотипирование (SNParray).
Запись результатов молекулярного кариотипа (ISCN 2016): arr[CRCh37]7q11.22q11.23(71997191_72310480)×3,18p11.32p11.21(136226_15154053)×1. Молекулярный анализ показывает, что в случае 14 утрачено практически всё короткое плечо хромосомы 18 (более 15 млн пар нуклеотидов). В полученных данных невозможно выявить критические участки, где локализованы гены, контролирующие синдром 18р-. Биоинформатический анализ выявил гены, связанные с отдельными клиническими признаками. В основном, это следующие гены: AFG3L2, LPIN2, MC2R, SBDSP1, частично ассоциированные с синдромом 18р-.
В результате молекулярного кариотипирования у данного пациента (табл. 1, случай 14) была не только подтверждена делеция 18р, но и выявлена микродупликация хромосомы 7, что, несомненно, внесло свой патологический вклад в клиническую картину пробанда. Известно, что размер всего короткого плеча хромосомы 18 – 16000000-16500000 пар нуклеотидов, включая гетерохроматин центромеры. В коротком плече хромосомы 18 содержится 118 генов, 57 из которых индексированы в базе данных OMIM. Выявленные у ребёнка гены лишь частично ассоциированы с клинической картиной. Необходимы дальнейшие молекулярно-цитогенетические исследования данного синдрома (18р-) с целью определения критических участков.
Результаты наших исследований демонстрируют, что (1) тяжесть клинических проявлений у пациентов с данным синдромом могут сильно варьировать в зависимости от точек разрыва при формировании делеции и, соответственно, от размера делетированного участка и локализованных в нём генов; (2) у пациентов с делецией 18р могут быть геномные аномалии, связанные с другими хромосомами, которые выявляются молекулярно-цитогенетическими методами (FISH, молекулярное кариотипирование), и эти аномалии могут; несомненно, вносить свой патологический вклад в клиническую картину; (3) необходимы дальнейшие молекулярно-цитогенетические исследования делеции короткого плеча хромосомы 18 для определения критических участков при синдроме 18р-.
Заключение. Исследования синдрома 18р- показывают, что эта хромосомная аномалия клинически связана, прежде всего, с возможной аномалией мозга и с различными формами умственной отсталости у детей, а также с ВПР и МАР. При диагностике этого заболевания необходимо применение современных молекулярно-цитогенетических методов исследования. Возможные аномалии генома часто имеют небольшие размеры, не позволяющие увидеть их при стандартном кариотипировании, но они могут значительно влиять на клиническую картину, а в сочетании с другими нарушениями могут увеличить тяжесть патологических проявлений, что необходимо знать для корректного эффективного медико-генетического консультирования семьи, т.е. речь идёт о персонализированном подходе к обследованию больного ребёнка. При этом диагностические технологии данной патологии и возможности применения молекулярно-цитогенетических подходов к исследованию синдрома важны в изучении корреляций фенотип-генотип [14, 19, 21, 22, 25].
Проведение цитогенетических и молекулярно-цитогенетических исследований данной хромосомной патологии не только позволит точнее охарактеризовать данный синдром, но и определить молекулярные механизмы патологических процессов, связанных с умственной отсталостью и пороками мозга, выявить критические участки синдрома делеции (частичной моносомии) короткого плеча хромосомы 18.
Информация о финансировании
Работа была частично поддержана грантом РФФИ и CITMA в соответствии с исследовательским проектом № 18-515-34005, ГОСЗАДАНИЕМ Минздрава России №. 121031000238-1, а также Правительственным заданием Министерства науки и высшего образования России №. AAAA-A19-119040490101-6
Благодарности
Авторы выражают благодарность с.н.с. О.С. Куринной, лаборантам-исследователям Н.С. Якушеву и В.Ю. Юровой за техническую помощь в подготовке статьи к публикации.














Список литературы