
Essentially pure partial trisomy 6(p21.31-p25) (case report and literature review)
Aннотация
Background: In reviewed literature, several patients with duplication or partial trisomy of the 6p region have been described. Most of these cases are associated with a partial monosomy of another chromosome. It has been suggested that partial trisomy 6p constitutes a well-defined syndrome. The aim of the study: To achieve a better clinical delineation of the 6p syndrome through the description of a patient with partial trisomy 6(p21.31-p25) comparing his characteristics with international reports and to discuss aspects of the phenotype of this syndrome. Materials and methods: A detailed clinical analysis of the patient’s condition was performed. The chromosomes were studied through the GTG-banding analysis. Results: On clinical examination we observed: a small anterior fontanel; fine, sparse and very pale hair, almost white hair; very white, translucent and thin skin; pale and sparse eyebrows and eyelashes; very narrow palpebral fissures with palpebral ptosis (blepharophimosis); a high nasal bridge, and straight nose with tiny nostrils; low implantation of the ears; microcephaly, neurodevelopmental and psychomotor delay; long philtrum, thin lips, the upper lip almost inverted and the mouth is small. From the neurological point of view there was evidence of trunk hypotonia and limb hypertonia. These are all typical features of trisomy 6p syndrome. A cytogenetic study of the patient and his father showed that trisomy 6p was due to an adjacent segregation I in paternal gametogenesis as the father is a 6,16-translocation carrier. Conclusion: The possible critical region is difficult to determine due to the clinical heterogeneity present in this syndrome. However, this case should be analyzed by molecular methods to determine more precisely the extent of the area involved in the trisomy.
К сожалению, текст статьи доступен только на Английском
Introduction. Several patients with duplication or partial trisomy of the 6p region have been described in consulted literature. Most cases were associated with a partial monosomy of another chromosome (product of an adverse segregation due to a translocation), so it has been suggested that partial trisomy 6p constitutes a well-defined syndrome [1-4]. When this trisomy appears in association with a partial monosomy of another chromosome, it is difficult to fully define whether the clinical features observed are actually due to trisomy 6p or to haploinsufficiency of the genes involved in the partial monosomy of a particular chromosome involved in the translocation. There are reports of this syndrome with a different extension of the trisomic segment on the short arm of chromosome 6, which also makes it difficult to determine exactly whether the clinical manifestations are due to a large clinical heterogeneity of the disease or to the different genes involved in partial trisomy [5].
Among the affections usually reported in partial trisomy 6p we can find a neurodevelopmental and psychomotor delay and other anomalies, such as severe or moderate dysmorphic features, low-set ears, prominent forehead, blepharophimosis, choanal atresia, arched palate, craniosynostosis, thin lips and tiny nostrils. Other findings have also reported cardiac defects, palpebral ptosis, intellectual disability, feeding problems, immunodeficiency, renal anomalies and pigmentary skin anomalies [1-11].
The current paper presents a partial trisomy 6p21.3-6p25 product of a 6;16 translocation, with the peculiarity that in chromosome 16 only the telomeric region (16q24) is involved, which implies that a possible monosomy of this region in the propositus does not represent a relevant clinical repercussion. Based on the particularities of this rearrangement, with an essentially pure trisomy 6p, we discuss the clinical features of the patient and compare it with a review of the literature in order to achieve a better clinical delineation of this syndrome and to discuss aspects of its phenotype.
Materials and methods. Karyotyping was performed using lymphocyte culture without exogenous serum and GTG bands at a resolution of 550 bands, according to standard laboratory techniques. The working algorithm described in [12] was applied. The software Metasystem was used for image capture, processing and analysis. Images were obtained by bright-field microscopy (Olympus BX-51).
Results and discussion. The patient is a 5-year-old boy, with non-consanguineous parents (a 22-year-old mother and a 27-year-old father), referred from the William Soler pediatric hospital. The proband showed a family history of mother in remission of Hodgkin's lymphoma, who concluded the treatment 4 and a half years before becoming pregnant, and maternal grandmother who died of cervical cancer. The child was born preterm at 36 weeks, by physiological delivery, with a weight of 1870g (less than the 5th percentile), supine length 44.5cm (less than the 5th percentile) and head circumference 30.5cm. The fetus was assessed as symmetrical intrauterine growth retarded. In the neonatology service, abdominal and trans-fontanel ultrasound and echocardiogram did not show visceral congenital defects.
Physical examination by a genetics specialist at 2 months of age showed: a small anterior fontanel; thin, sparse and very pale hair, almost white; very white, translucent and thin skin; pale and sparse eyebrows and eyelashes. Very narrow palpebral fissures with palpebral ptosis (blepharophimosis), a high nasal bridge, a straight nose with tiny nostrils, low implantation of the ears, long philtrum, thin lips, the upper lip almost inverted and the mouth is small. In the thorax teletelia is detected. In the hands and feet there were deep palmar and plantar folds. From the neurological point of view there was evidence of trunk hypotonia and limb hypertonia.
Follow-up of the patient revealed the presence of other disorders such as gastroesophageal reflux, irritability, neurodevelopmental and growth retardation and microcephaly. Neuroimaging studies of brain structures showed signs of brain atrophy, predominantly left temporal.
At the present moment, the child is 5 years old and has presented some changes in the phenotype, he maintains short stature and similar facial features, but the hair has taken a reddish and tarnished appearance, with a thicker and rougher consistency. From the psychomotor point of view, the delay in the acquisition of skills persists and, fundamentally, a significant speech proficiency delay. Hearing loss has been ruled out as a cause of this disability.
Cytogenetic analysis
Fifteen metaphases were observed under the bright-field microscope and at least 5 karyotypes were analyzed.
The 15 metaphases showed a male karyotype with an apparent addition on the long arm of chromosome 16 in all metaphases studied: 46, XY, add, (16), (q23) (Figure 1).
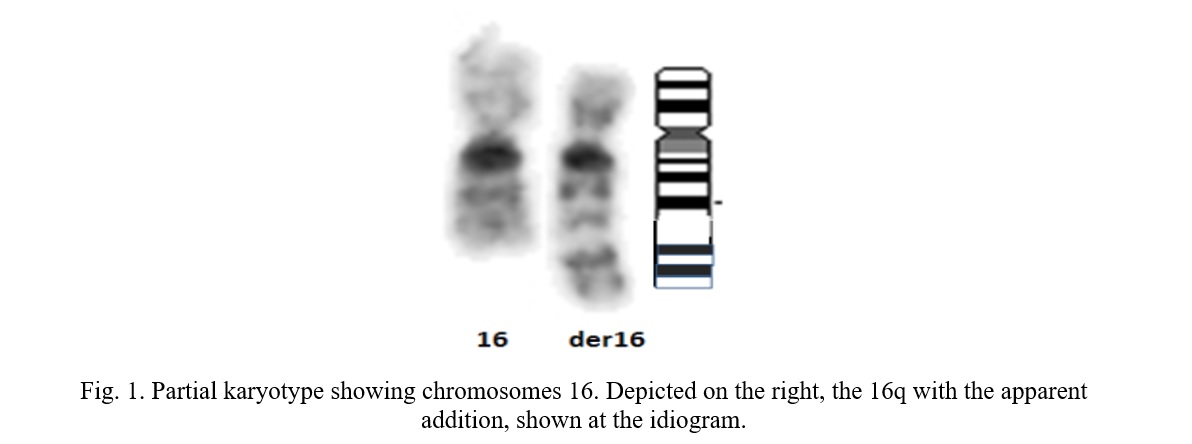
To determine the inherited or de novo possible origin of this rearrangement, a chromosomal study of the parents was indicated. The father was found to carry a translocation between the short arm of chromosome 6 and the long arm of chromosome 16, as shown in Figure 2.
Karyotype: 46, XY, t (6,16), (p21.31, q24). Chromosomal analysis of the mother showed a normal 46, XX karyotype.

The short arm of chromosome 6 has 1 204 genes making it difficult to estimate the contribution of gene expression to the trisomy 6p phenotype. [13] The diversity of phenotypic features that patients with the partial trisomy 6p may present should be considered. However, unusual facial features, a developmental delay, a prominent forehead, microcephaly, blepharophimosis and palpebral ptosis, [1-11] can also be found in other patients with completely different chromosomal rearrangements. Furthermore, if we contemplate that not all patients with trisomy 6p present all the phenotypic features described above, it becomes difficult to consolidate a phenotypic pattern that characterizes trisomy 6p as a syndrome.
The case described in this report presents a partial trisomy involving the 6p21.31-p25 region and the clinical features most commonly reported in the syndrome are summarized in Table 1. The theoretical analysis of the region with partial trisomy 6p allows us to point out which genes may be responsible for the clinical features of the patient hereby described. The craniofacial alterations link to the BMP6 gene (6p24.3) has been suggested by Castiglione et al. 2013 [13, 14]. The bone morphogenetic proteins (BMPs) are a family of secreted signaling molecules that can induce ectopic bone growth. Many BMPs are part of the transforming growth factor-β superfamily [15, 16]. A triple dose of this gene could be responsible for malformations such as craniosynostosis, choanal atresia and other more moderate cranial malformations. This gene is within the region implicated in the partial trisomy 6p of the described patient, however, neither choanal atresia nor craniosynostosis are observed in this patient. Similar situations are reported in the literature by several authors. Refer to Table 1.
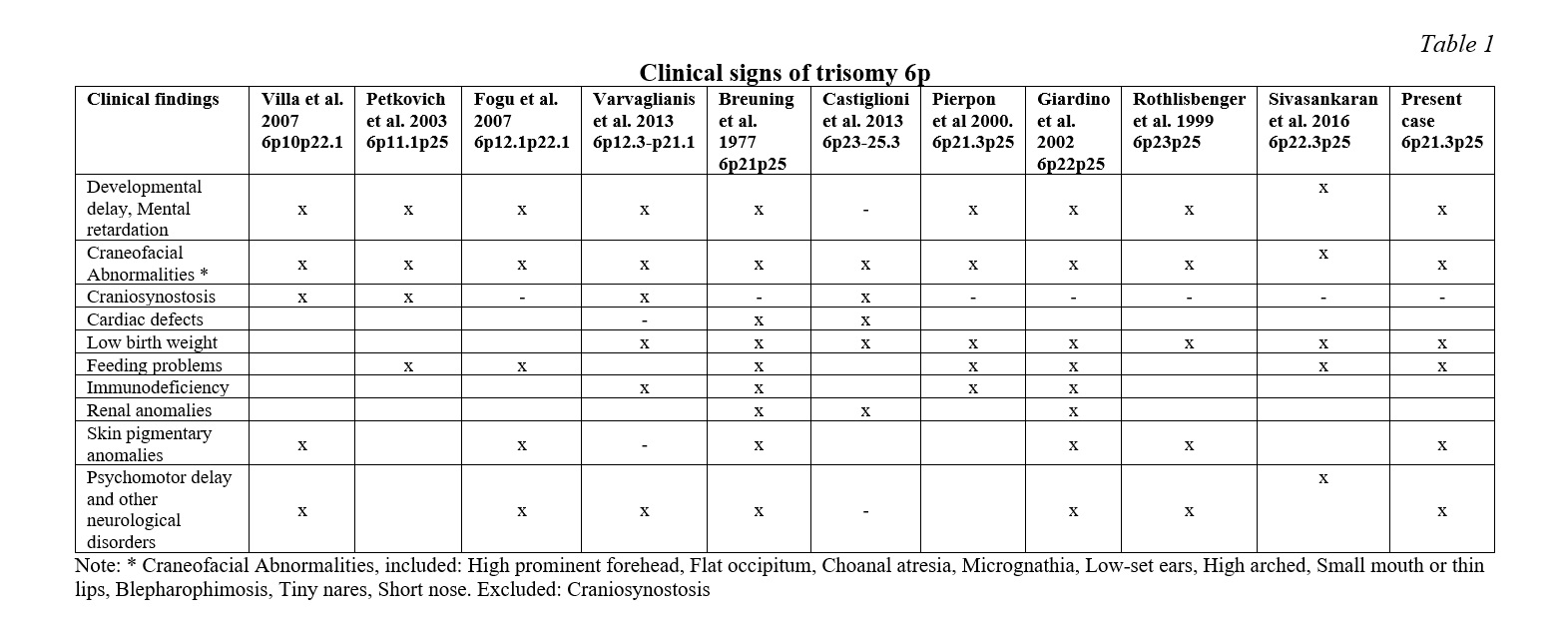
As suggested by Varvagiannis et al, 2013 [17] craniosynostosis is caused by a triple dosage of the RUNX2 gene (6p21.1). The RUNX2 gene (RUNX family Transcription factor 2) is a member of the RUNX family of genes that are essential for osteoblastic differentiation and skeletal morphogenesis. RUNX2, also known as CBFA1, maps to 6p21 and encodes the RUNT-related transcription factor 2, a master regulator of osteoblast differentiation. Our cytogenetic analysis of the patient under study excludes the 6p21.1 region. In addition, molecular methods were not used in our analysis of the rearrangement breakpoints. The clinical description of this patient does not report craniosynostosis, which suggests that the RUNX2 gene is outside of the partial trisomy region despite the relative proximity to the breakpoint (6p21.31) involved in the translocation [6, 16]. Villa et al. hypothesized that duplication of the gene for bone morphogenetic protein 5 (BMP5) might be responsible for the premature fusion of the patient’s sagittal sutures, but this gene is located at 6p12.1 completely outside the trisomy 6p region of the proband.
Regarding the ocular anomalies present in the reported case (blepharophimosis, palpebral ptosis, epicanthal folds) Su and collaborators (2012) [18] suggested nine genes that could be involved in these anomalies: FOXQ1(6p25. 3), FOXF2(6p25.3), FOXC1(6p25.3), NRN1(6p25.1), EDN1(6p24.1), ATXN1(6p22.3), DEK (6p22.3), E2F3(6p22.3) and NRSN1(6p22.3). The FOX genes have also been considered by other authors to be responsible for the ocular malformations in these syndromes [19]. For example, the FOXC1 gene plays a fundamental role in the regulation of embryonic and ocular development; mutations in this gene could cause different types of glaucoma and iris dysgenesis [13].
In a general sense many of the phenotypic characteristics of patients with partial trisomy at 6p are attributable to the 6p25-p21 region; among them: pre- and postnatal growth retardation, microcephaly, a prominent forehead, ocular malformations, low ear implantation, long philtrum, hypoplastic kidneys, congenital heart defects, recurrent infectious episodes [15]. Many of these conditions are present in the reported case and coincidentally the region with trisomy 6p21.31-p25 encompasses the region described previously in the propositus.
The patient has a moderate mental disability, a frequent characteristic of trisomy 6p, even though in some cases the mental disability is mild and allows normal social interaction [7, 20, 21]. The most remarkable affection in this patient is the speech proficiency delay at 5 years of age. Polymorphisms and CNVs (copy number variations) in several of the genes within the 6p21.3-6pter region (ATXN1, DTNBP1, JAR1D2, LYRM4, MYLIP, NQO2, NRN1, RREB1, RIPK1, SERPINB1) have been reported to be implicated in intellectual disability [21-31].
As previously mentioned, one of the limitations of present study is that the chromosomes breakpoints were not determined by molecular methods. Regarding the q24 breakpoint on chromosome 16 and based on the child's clinical findings we found no phenotypic alterations corresponding to a possible monosomy of 16q24.1 or 16q24.3, the most likely sites to be involved in the rearrangement. Monosomy at 16q24.3 is associated with seizures and autism spectrum, brain abnormalities and neonatal thrombocytopenia. Monosomy at 16q24.1, on the other hand, is associated with lethal lung disease, with refractory pulmonary hypertension and the child dies in the first months of life [32, 33]. None of these alterations corresponds to the clinical findings of the proband. It follows that the part of chromosome 16q involved in the 6,16 translocation is the telomere of chromosome 16, a highly repetitive, non-coding region of DNA.
In a general sense, this can be considered an essentially pure 6p21.31-p25 trisomy, because the found monosomy suggests a non-coding region on chromosome 16. All the clinical features reported in the patient correspond to trisomy of the 6p25-p21.31 region. Other authors consider that genes located between 6p25.1 and 6p25.2 are responsible for the clinical features of this trisomy, 13 which is not consisting with our finding. Villa et al. report a case with typical features of the syndrome in which band 6p25 trisomy is not involved [8].
The careful clinical delineation of the patient combined with the chromosomal findings and the international literature reports suggest that the critical region fundamental to this syndrome cannot only be circumscribed to 6p25, as it is probably more extensive. However, this case should be analyzed by molecular methods to determine more precisely the extent of the area involved in the trisomy. In addition, a detailed molecular characterization of the genes in this region and their function during the ontogeny of these patients affected with this trisomy is necessary, because some of the features of this syndrome, such as heart disease and renal anomalies, are not present in the proband. On the other hand, triple dose genes, BMP6, do not always affect the phenotype of individuals with trisomy 6p in the same way, which could suggest a variable expressiveness of this gene in the clinic of this syndrome.














Список литературы