
Роль метаболизма кортизола в реализации патогенетических звеньев развития остеопороза – обоснование поиска новых фармакотерапевтических мишеней (обзор)
Aннотация
Актуальность: Остеопороз является важной медицинской и социальной проблемой общественного здравоохранения в стареющем или пожилом обществе. Остеопороз вызывается дисбалансом в костном ремоделировании, которое представляет собой непрерывный процесс разрушения зрелой костной ткани остеокластами (резорбция кости) и формирования новой костной ткани остеобластами (образование кости). Система костного гомеостаза, регулирующая функциональную активность остеокластов и остеобластов представлена широким спектром молекул. Достигнутое сегодня понимание молекулярных механизмов костного гомеостаза позволяет существенно изменить и расширить парадигмы лечения и профилактики остеопороза. Цель исследования:Рассмотреть основные патогенетические пути, через которые реализуется влияние системы метаболизма кортизола на развитие остеопороза и обозначить пути поиска новых терапевтических подходов к лечению и профилактике обозначенной патологии. Материалы и методы:Для достижения поставленной цели был проведен анализ литературных источников по проблеме влияния метаболизма кортизола на развитие остеопороза, опубликованных за последние 10 лет. Результаты:На сегодняшний день в литературе имеются весомые предпосылки прямой связи нарушений метаболизма стероидных гормонов с развитием остеопороза и нарушением остеорепаративных процессов. В настоящем литературном обзоре представлены основные патогенетические пути, обуславливающие процессы, ведущие к снижению плотности костной ткани при нарушениях метаболизма кортизола. Фермент 11b-гидроксистероиддегидрогеназа (11b-HSD), представленный двумя изоформами осуществляет взаимное превращение кортизона и кортизола в тканях. С использованием методов обратной генетики были установлены системные последствия нокаута обеих изоформ. Убедительные доказательства демонстрируют, что оба фермента вовлечены в патогенез остеопороза. Поскольку животные с дефицитом 11b-HSD 2 типа характеризуются провоспалительной активацией эндотелия мы предполагаем, что отдельный интерес представляет дальнейшее изучение взаимодействия между эндотелием и костной тканью. Заключение:Эффекты глюкокортикоидов на экспрессию eNOS, по-видимому, существенно модулируется изоферментами 11β-HSD. Установленная связь между 11β-HSD и NO может рассматриваться перспективная фармакотерапевтическая мишень. В этой связи, фармакотерапевтический подход, направленный на восстановление баланса оксида азота в костной и эндотелиальной тканях, рассматривающийся в настоящее время как один из наиболее актуальных способов коррекции остеопороза может быть актуальным и при нарушении обмена кортизола вследствие недостаточности 11β-HSD 2.
Введение. Остеопороз – заболевание, характеризующееся снижением костной массы, ухудшением состояния костной ткани и нарушением микроархитектоники кости. От остеопороза страдают более 75 миллионов человек в Европе, Японии и США, остеопорозом обусловлено ежегодно более 2,3 миллионов переломов в указанных странах [1, 2]. Остеопороз называют «молчаливой эпидемией», потому что во многих случаях заболевание на первых стадиях никак себя не проявляет. Всемирная организация здравоохранения официально определила остеопороз (ОП) как одно из десяти важнейших хронических заболеваний человечества, поскольку он очень широко распространен, имеет четкое определение, методы диагностики, и возможности для профилактики и лечения. ОП – системное заболевание скелета из группы метаболических остеопатий – характеризуется уменьшением костной массы и нарушением микроархитектоники костной ткани, что приводит к снижению прочности кости и, как следствие, к повышению риска возникновения переломов. Будучи одной из наиболее частых причин патологических инвалидизирующих переломов, остеопороз существенно ограничивает качество и продолжительность жизни и усугубляет течение сопутствующих заболеваний [3, 4].
Остеопороз вызывается дисбалансом ремоделирования кости, который представляет собой непрерывный процесс, при котором зрелая костная ткань удаляется остеокластами (резорбция кости), а новая костная ткань формируется остеобластами (образование кости). Чрезмерная резорбция кости или недостаточное образование новой кости в процессе костного ремоделирования может привести к остеопорозу [5].
Научные достижения современной биомедицины привели к прорыву в терапии больных со снижением плотности костной ткани. За последние три десятилетия арсенал средств для лечения подобных пациентов продвинулся от заместительной терапии эстрогенными препаратами и витамином D до применения таких революционных средств как бисфосфонаты, ралоксифен (селективный модулятор рецептора эстрогена), деносумаб (антитело к внутриклеточному фактору RANKL) и рекомбинантный паратиреоидный гормон. Тем не менее, значительный прогресс в фармакотерапии остеопороза не снижает актуальность поиска новых остеопротекторов и мишеней для воздействия на процессы остеоремоделирования. Актуальность дальнейших исследований продиктована, в числе прочего, тем, что для успешной персонализированной терапии врач должен располагать большим выбором резервных стратегий лечения. Например, одобренный FDA в первой половине 2019 года препарат ромосозумаб, проходил долгую процедуру регистрации из-за высокого риска сердечно-сосудистых осложнений. Кроме того, при применении некоторых современных антиостеопоротических препаратов происходит развитие толерантности.
Известно, что снижение остеорепаративных процессов и увеличение остеорезорбции часто связаны с нарушением обмена стероидных гормонов. В связи с этим, основными факторами риска для развития остеопороза является постменопаузальный период и длительная терапия глюкокортикоидами [6]. Поэтому одним из наиболее очевидных направлений для поиска молекулярных предикторов и новых мишеней для фармакотерапии остеопороза является система тканевого метаболизма стероидных гормонов. Скелет является одной из классических мишеней глюкокортикоидных гормонов. Кортикостероидная активация при гиперкортицизме, длительном лечении кортикостероидными гормонами или альдостеронизме ассоциирована со снижением плотности костной ткани [7, 8].
Фермент 11b-гидроксистероиддегидрогеназа (11b-HSD), представленный двумя изоформами осуществляет взаимное превращение кортизона и кортизола в тканях. С использованием методов обратной генетики были установлены системные последствия нокаута обеих изоформ. Убедительные доказательства демонстрируют, что оба фермента вовлечены в патогенез остеопороза, при этом 11b-HSD 2 типа по всей видимости выполняет остеопротективную роль. Тем не менее, на сегодняшний день отсутствуют комплексные in vivo и in vitro исследования, позволяющие окончательно судить о его вкладе в остеопороз. Кроме того, поскольку животные с дефицитом 11b-HSD 2 типа характеризуются провоспалительной активацией эндотелия мы предполагаем, что отдельный интерес представляет изучение взаимодействия между эндотелием и костной тканью. Ввиду анатомических особенностей строения сосудистого русла скелета, функция эндотелия является критичной для гомеостаза костной ткани. Предыдущие исследования нашего коллектива подтвердили эту связь и продемонстрировали, что восстановление функции эндотелия, в том числе NO-продуцирующей обладает высоким терапевтическим потенциалом при коррекции экспериментального остеопороза, а анализ и оценка новых физиологических мишеней для фармакологической коррекции нарушений процессов костного ремоделирования является актуальной научной задачей.
Цель исследования. Рассмотреть основные патогенетические пути, через которые реализуется влияние системы метаболизма кортизола на развитие остеопороза и обозначить пути поиска новых терапевтических подходов к лечению и профилактике обозначенной патологии.
Материалы и методы исследования. Для достижения поставленной цели был проведен анализ литературных источников по проблеме влияния метаболизма кортизола на развитие остеопороза, опубликованных за последние 10 лет.
Остеопороз и метаболизм стероидных гормонов
Плейотропность биологических эффектов стероидных гормонов и сложность путей их метаболизма обуславливают трудности в окончательном понимании патогенетических аспектов развития и прогрессирования остеопороза. В тканях, экспрессирующих рецепторы к стероидным гормонам, функционирует сложный каскад энзиматичеких превращений, который лимитирует взаимодействие гормонов с их внутриклеточными лигандами [9].
Обнаружение рецепторов стероидных гормонов на остеоцитах привело ученых к мысли, что стероидные гормоны должны модулировать их биосинтетическую активность. Вскоре после этого было показано, что стероидные гормоны не просто влияют на функциональное состояние существующих остеоцитов, но фактически являются мощными регуляторами их образования и продолжительности жизни. В 1992-1995 годах показаны цитокин-опосредованные механизмы регуляции остеокластогенеза эстрогенами и андрогенами [10, 11]
Среди ключевых ферментов, влияющих на кинетику стероидов в тканях можно выделить две изоформы 11β-гидроксистероиддегидрогеназы (11β-HSD), осуществляющие пререцепторный контроль биологической активности кортизола. 11β-HSD 1 типа осуществляет катализ метаболитов глюко- и минералокортикоидов в их активные формы, а 11β-HSD 2 типа, напротив, приводит к инактивации стероидов, предупреждая избыточную активацию минералокортикоидных и кортикостероидных рецепторов. У человека и грызунов две изоформы, кодируются разными генами, расположенными на разных хромосомах, и баланс между их активностью определяет индивидуальный ответ ткани на гормональные и паракринные воздействия [12, 13].
Из актуальных работ, показывающих роль метаболизма стероидных гормонов в развитии и дифференцировке костной ткани мы выделяем работы группы ученых под руководством Hao Xiao, показавших что низкий уровень экспрессии 11β-HSD 2 опосредует предрасположенность к остеопорозу, вызванному пренатальным воздействием кофеина у крыс-самцов [14], а также исследование группы американских ученых, показавших, что Hsd11b1 является мишенью для разработанного ими лекарственного средства, лимитрирующего остеогенез и функциональную активность отсеокластов [15].
Вызванная глюкокортикоидами активация остеокластов и инактивация остеобластов приводит к отрицательному балансу кальция в костях. Блокируя в остеобластах Wnt-сигнализацию и экспрессию транскрипционного фактора Runx2, глюкокортикоиды останавливают остеогенез и остеорепарацию [7]. Работы в данном направлении закономерно привели к пониманию важной роли тканевых ферментов метаболизма стероидов в развитии остеопороза. В костной ткани человека и грызунов присутствуют оба фермента 11β-HSD, однако изоформа 1 экспрессируется только в остеобластах и остеокластах, а изоформа 2 – только в остеобластах [16, 17]. В исследовании Arampatzis S. et al. (2013) доказано обоснованное предположение, что дисбаланс активности изоформ 11β-HSD приводит к накоплению кортизола в костной ткани и стимуляции активности остеокластов с дальнейшей деминерализацией костной ткани [18].
Мыши с нокаутом 11β-HSD 1, созданные и охарактеризованные в Эдинбурге под руководством Юрия Котелевцева в 1999 г. демонстрируют метаболическую толерантность к высоким дозам глюкокортикоидов, что логично истекает из функции данного фермента [19]. Недавно было проведено исследование, подтверждающее, что данная линия устойчива к развитию остеопороза при воздействии высоких доз глюкокортикоидов [20]. Мыши с нокаутом 11β-HSD 2 типа, полученные тем же коллективом, склонны к гипертензии, сердечной недостаточности, гипокалиемии и развивают клиническую картину, схожую с синдромом гиперальдостеронизма [21, 22]. Однако, несмотря на глубокий фенотипический анализ, проведенный за 20 лет, сведения о состоянии костной ткани у данной линии практически отсутствуют. Наиболее близкой к этому направлению работой было создание в 2004 г. линии, с экспрессией 11β-HSD 2 в остеокластах, которая показала устойчивость к остеопорозу при введении высоких доз глюкокортикоидов [23, 24]. Также в 2017 году вышла работа, где продемонстрировано, что гиперэкспрессия 11β-HSD 2 в остеобластах, привела к снижению экспрессии проапоптических факторов Fas и каспазы-8 in vitro [25].
Эндотелий опосредованная активация процессов костной резорбции
Существуют и другие предпосылки, указывающие на тесную связь между активностью 11β-HSD 2 и минеральной плотностью костной ткани. Ряд исследований демонстрирует, что нокаут HSD11B2 приводит к существенному ухудшению функции сосудистой стенки, что выражается в прогрессировании атеросклероза и увеличении экспрессии молекул межклеточной адгезии эндотелиоцитами [26]. Последнее обстоятельство представляется нам особенно важным ввиду тесной связи между функцией эндотелия и морфофункциональным состоянием костной ткани, сосудистая сеть которой представлена только эндотелиальным монослоем.
С использованием молекулярно-биологических подходов и методов обратной генетики в настоящее время установлена роль обоих ферментов в регуляции артериального давления, электролитного состава плазмы крови, влиянии на минеральный обмен костной ткани и кардиотропное действие.
Кроме того, поскольку животные с дефицитом 11b-HSD 2 типа характеризуются провоспалительной активацией эндотелия [26], мы предполагаем, что отдельный интерес представляет изучение взаимодействия между эндотелием и костной тканью. Ввиду анатомических особенностей строения сосудистого русла скелета, функция эндотелия является критичной для гомеостаза костной ткани. Предыдущие исследования нашего коллектива подтвердили эту связь и продемонстрировали, что восстановление функции эндотелия, в том числе NO-продуцирующей обладает высоким терапевтическим потенциалом при коррекции экспериментального остеопороза, а анализ и оценка новых физиологических мишеней для фармакологической коррекции нарушений процессов костного ремоделирования является актуальной экспериментальной задачей. Терапевтический потенциал подобного подхода был продемонстрирован с использованием гипоэстроген-индуцированного и ассоциированного с эндотелиальной дисфункцией остеопороза [27, 28]. В связи с приведенной информацией, мы считаем обоснованной гипотезу о тесной связи фермента 11β-HSD 2 с остеопорозом, нарушением взаимодействия между эндотелием и костной тканью, а также снижением биосинтеза NO.
Несмотря на доказательства участия ферментных систем 11β-HSD 1 и 2 в регуляции функции эндотелия и минеральный обмен костной ткани, на сегодняшний день отсутствуют данные, характеризующие процессы костного ремоделирования и остеорепарации у мышей с генотипом 11β-HSD2–/– и 11β-HSD2+/–. Отдельные предпосылки, указывающие на тесную связь данного фермента и функциональное состояние эндотелиоцитов, обуславливают целесообразность комплексного целенаправленного изучения взаимодействия костной ткани и сосудистого эндотелия с оценкой роли оксида азота в реализации остеотропных эффектов 11β-HSD 2.
Остеопороз и сахарный диабет – роль метаболизма кортизола
Как и остеопороз, сахарный диабет является современной пандемией со значительной заболеваемостью и смертностью [29]. При хроническом течении СД оказывает неблагоприятное воздействие на различные органы и ткани, включая кости, нервы, мышцы, сетчатку, сердечно-сосудистую систему и почки [30].
С точки зрения рассматриваемых в данной статье механизмов реализации патогенетических звеньев развития остеопороза основной интерес представляют пути, через которые реализуется влияние повышенных концентраций кортизола на метаболизм глюкозы. Повышение экспрессии 11β-HSD-1 под действием высоких концентраций глюкозы закономерно приводит к увеличению концентрации кортизола. Известно, что у тучных людей в жировой ткани повышена экспрессия 11β-HSD-1 [31]. В тоже время показано, что трансгенные мыши, селективно сверхэкспрессирующие 11β-HSD1 в жировой ткани, характеризуются выраженным метаболическим синдром с висцеральным ожирением, дислипидемией, инсулинрезистентным диабетом и гипертонией [32, 33].
Кортизол регулирует метаболизм и дифференцировку адипоцитов, способствуя адипогенезу и увеличению запасов висцерального жира [34, 35, 36]. Также кортизол уменьшает чувствительность тканей к инсулину снижая сродство рецепторов, или даже количество рецепторов к инсулину, снижает поглощение глюкозы клеткой за счет изменения транслокации GLUT4 на плазматическую мембрану [37]. В 2010 году показана клиническая эффективность ингибитора 11β-HSD1 под шифром INCB13739 в контроле гипергликемии и улучшении чувствительности к инсулину у пациентов с сахарным диабетом, плохо контролируемым метформином [38].
Влияние глюкокортикоидов на секрецию инсулина заключается в снижении секреции инсулина β-клетками поджелудочной железы, изменении ими окислительного метаболизма глюкозы, а также активации реполяризации К+-каналов. Указанные процессы индуцируют образование активных форм кислорода, повышают активность 11β-HSD1 и снижают эффективность внутриклеточных ионов Ca2+ в секреторном ответе [39]. Со снижением функции β-клеток хроническая гипергликемия вызывает окислительный стресс, воспаление, выработку активных форм кислорода (АФК) и конечных продуктов гликирования (КПГ), вызывая повреждение органов и снижение прочности костей. В частности, накопление КПГ в диабетическом костном коллагене определяет снижение свойств коллагена и повышенную склонность к переломам. КПГ и гипергликемия также напрямую ингибируют образование кости посредством подавления функции остеобластов. При сахарном диабете 1-го типа (СД1) и на поздних стадиях сахарного диабета 2-го типа (СД2) костеобразование также снижается из-за дефицита инсулина за счет ингибирующего действия на остеобласты либо непосредственно, либо через изменения в инсулиноподобном факторе роста 1. Изменения в метаболическом пути кальций-паратиреоидный гормон (ПТГ) приводят к отрицательному балансу кальция, тем самым способствуя деминерализации костей при сахарном диабете [40]
Эстрадиол-опосредованная регуляция активности 11b-HSD
Остеопороз является серьезной проблемой для здоровья женщин в постменопаузе. Костная масса увеличивается в детстве, достигая пика к третьему или четвертому десятилетию жизни женщины. После этого начинается потеря костной массы, которая ускоряется в период менопаузы. В исследованиях сообщается, что частота остеопороза удваивается примерно каждые 5 лет, начиная с возраста 45-49 лет с 3,3% и постепенно увеличиваясь до 50,3% в возрасте 85 лет и старше [41, 42].
Известно, что ремоделирование кости осуществляется остеобластами, остеокластами и остеоцитами. Отрицательный дисбаланс ремоделирования кости, при котором резорбция кости превышает образование кости, приводит к остеопорозу. На клеточном уровне существует несколько механизмов нарушения костного ремоделирования и остеорепарации, связанных с дефицитом эстрогенов [41]. Рецепторы к эстрогену (ER) в высокой степени экспрессируются в остеобластах, остеокластах и остеоцитах, оказывая защитное действие на кость. Эстроген связывается с ER, которые регулируют экспрессию белков, кодирующих гены мишеней эстрогена - интерлейкин-1 (IL-1), инсулиноподобный фактор роста 1 (IGF1) и трансформирующий фактор роста бета (TGFβ) [43]. Недостаток эстрогена изменяет экспрессию генов-мишеней эстрогена, увеличивая секрецию ИЛ-1, ИЛ-6 и фактора некроза опухоли (ФНО). Исследования также показали, что дефицит эстрогена напрямую влияет на дифференцировку клеток и апоптоз [44].
Установлено, что эстрадиол подавляет активность 11β-HSD1 [45], что приводит к снижению образования активного кортизола на клеточном уровне. Используя дифференцированные адипоциты 3T3-L1 показано, что 17В-эстрадиол ингибирует активность 11β-HSD1. Антагонисты рецептора эстрогена, ICI-182 780 и тамоксифен, не смогли блокировать эффект его ингибирования. Кроме того, ацетилирование или α-эпимеризация 17-гидроксигруппы 17В-страдиола ослабляет его ингибирующее действие на 11β-HSD1. Кинетическое исследование показало, что 17в-эстрадиол действует как неконкурентный ингибитор 11β-HSD1 [36]. Кроме того, эстрогены также могут подавлять действие лиганда ядерного фактора-κβ (RANKL), ингибируя, таким образом, образование остеокластов и резорбтивную активность кости, реализуемую за счет кортизола [46].
Перспективные фармакотерапевтические мишени
Таким образом, на сегодняшний день в литературе имеются весомые предпосылки прямой связи нарушений в метаболизме кортизола с развитием остеопороза и нарушением остеорепаративных процессов. Основываясь на проведенном литературном обзоре, мы можем выделить основные патогенетические механизмы, связанные с метаболизмом кортизола и обуславливающие процессы, ведущие к снижению плотности костной ткани (Рис.).
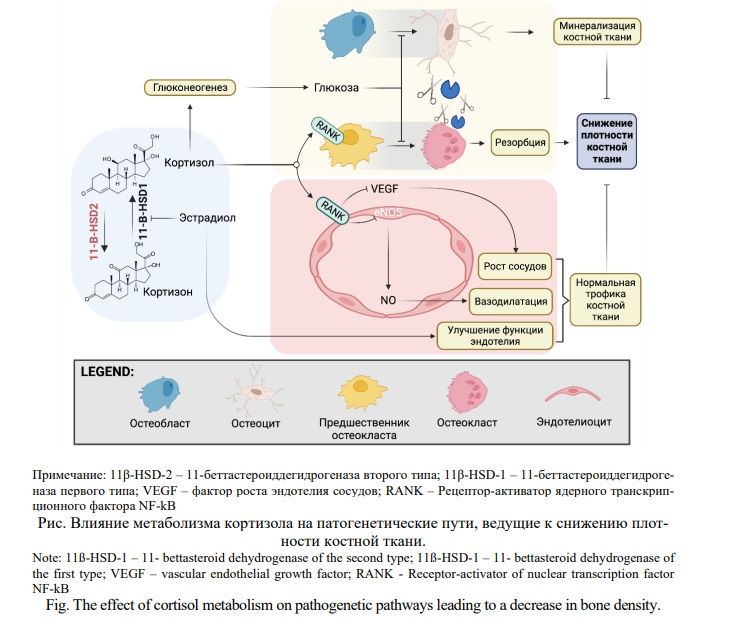
Высокие концентрации кортизола непосредственно влияют на лиганд-акцепторную систему RANK/RANKL/OPG путем увеличения экспрессии RANKL, активации RANK c дальнейшим увеличением интенсивности дифференцировки остеокластов. Также, высокие концентрации кортизола приводят к активации глюконеогенеза в печени что приводит к повышению концентрации глюкозы. Повышение концентрации глюкозы в свою очередь приводит к активации фермента 11β-HSD-1 и дальнейшему нарастанию концентрации кортизола. На сегодняшний день роль сахарного диабета в активации процессов костной резорбции и нарушении процессов остеорепарации не вызывает сомнений. Гипергликемия может вызывать остеопороз и патологическую хрупкость костей, воздействуя на костные и мышечные клетки различными возможными путями. Биомолекулярные основы развития остеопороза достаточно подробно изложены в ряде литературных обзоров [40, 47-50]. Эстрадиол реализует свое защитное действие путем влияния на метаболизм кортизола угнетая активность 11β-HSD-1 и улучшая функцию сосудистого эндотелия, что имеет особый эффект в костной ткани с учетом анатомических особенностей строения сосудов костей.
Поскольку в настоящее время нет эффективных способов фармакологически повышать активность 11β-HSD 2, особую значимость приобретает изучение метаболических и регуляторных путей, сопряженных с 11β-HSD 2. В этой связи отдельный интерес представляет связь 11β-HSD с метаболизмом оксида азота (NO) - фактора, в значительной степени влияющего на остеорепаративные процессы. Считается, что NO широко вовлечен в развитие, функционирование и ремоделирование скелета на всех этапах онтогенеза. В костной ткани обнаруживаются все три изоформы NO-синтаз (eNOS, iNOS и nNOS) [51]. При этом, как и в других тканях, сложно однозначно оценить эффекты оксида азота с точки зрения желательных или нежелательных. Например, остеопротективное действие эстрогенов тесно связано с их способность стимулировать синтез оксида азота в остеобластах. В то же время, остеопороз на фоне персистирующего системного воспалительного ответа также связан с повышением продукции оксида азота за счет NO-эргического действия цитокинов. Считается, что базальные концентрации NO обладают в отношении костной ткани регенеративным, а избыточные – ингибирующим эффектом. Исследования в области генетической эпидемиологии, выявившие ассоциацию полиморфных вариантов гена eNOS с риском развития остеопороза, дополнительно подтверждают эту связь [52, 53, 54]. В данном контексте важно, что гипертонический и другие эффекты глюкокортикоидов могут быть частично опосредованы подавлением экспрессии эндотелиальной синтазы оксида азота (eNOS). При этом эффекты глюкокортикоидов на экспрессию eNOS, по-видимому, существенно модулируется изоферментами 11β-HSD. Установленная связь между 11β-HSD и NO может рассматриваться перспективная фармакотерапевтическая мишень. Данные, полученные коллективом Юрия Котелевцева при фенотипировании мышей с дефицитом 11β-HSD 2 демонстрируют, что у них происходит снижение эндотелийзависимой вазодилатации, указывающее на подавление активности eNOS в эндотелии сосудов [55].
Заключение. Исходя из представленных литературных данных, очевидно, что обмен кортизола является одним из ключевых факторов, регулирующих процессы трофики, метаболизма и минерализации костной ткани, а высокие концентрации кортизола приводят к каскаду процессов, конечной точкой которых является снижение нормальной трофики костной ткани и остеопороз. Снижение концентрации кортизола возможно за счет уменьшения его синтеза с помощью блокады фермента 11β-HSD-1 или за счет ускорения процессов его метаболизма в неактивную форму (кортизон) стимулируя активность 11β-HSD-2. В этой связи, фармакотерапевтический подход, направленный на восстановление баланса активности ферментов 11b-гидроксистероиддегидрогеназы 1 и 2 типов может рассматриваться как один из наиболее актуальных способов коррекции предотвращения реализации патогенетических звеньев развития остеопороза, а ферменты 11β-HSD-1 и 11β-HSD-2 являются обоснованными фармакотерапевтическими мишенями, на которые может быть направлен поиск новых лекарственных средств для лечения остеопороза и нарушений костного ремоделирования.
Информация о финансировании
Исследование выполнено за счет гранта Российского научного фонда № 22-25-00376 (https://rscf.ru/project/22-25-00376).














Список литературы