
Взаимосвязь мобильных генетических элементов с некодирующими РНК в развитии атеросклероза (обзор)
Aннотация
Актуальность: Атеросклероз является ведущей причиной сердечно-сосудистой патологии взрослого населения всего мира. Ассоциация болезни с возрастом свидетельствует о наличии общих механизмов развития старения и атеросклероза. В научной литературе появляется все больше данных о роли некодирующих РНК и мобильных генетических элементов в механизмах старения и атеросклероза. Поиск молекулярных механизмов болезни на эпигенетическом уровне позволит разрабатывать новые методы терапии. Цель исследования: Определить роль транспозонов и некодирующих РНК в этиопатогенезе атеросклероза и их взаимосвязь между собой в данных процессах. Материалы и методы: Использованы базы данных Scopus, WoS, PubMed для анализа роли микроРНК, длинных некодирующих РНК, транспозонов в развитии старения и атеросклероза. Результаты: Согласно проанализированной литературе, важным фактором развития атеросклероза является патологическая активация транспозонов при старении, вызывающая интерфероновый ответ и асептическое воспаление в организме, в том числе в стенках сосудов. Определена роль эпигенетических факторов, в этиопатогенезе атеросклероза, включая микроРНК и длинные некодирующие РНК, которые изменяют экспрессию специфических генов в макрофагах, гладкомышечных клетках сосудов и эндотелиоцитах. Активация транспозонов отражается в изменении экспрессии произошедших от них в эволюции или образующихся при процессинге их транскриптов длинных некодирующих РНК и микроРНК. Анализ научной литературы позволил обнаружить 64 произошедших от транспозонов микроРНК, изменения экспрессии которых характерны для атеросклероза. Из 64 выявленных микроРНК 34 ассоциированы со старением, что свидетельствует о роли патологически активированных при старении транспозонов в инициации развития атеросклероза. Заключение: Поскольку транспозоны являются драйверами эпигенетической регуляции в онтогенезе, полученные результаты впервые в научной литературе описывают наиболее вероятные механизмы влияния механизмов старения на развитие атеросклероза на эпигенетическом уровне. Это обусловлено патологической активацией транспозонов с возрастом, что оказывает влияние на изменение экспрессии произошедших от них некодирующих РНК за счет наличия комплементарных последовательностей и участия в общих эпигенетических сетях регуляции функционирования генома. Полученные данные о роли произошедших от транспозонов микроРНК в развитии как атеросклероза, так и старения, подтверждают предложенные механизмы патогенеза болезни.
Введение. Атеросклероз (АС) является ведущей причиной сердечно-сосудистых заболеваний в мире. Его основные клинические проявления включают ишемическую болезнь сердца и головного мозга и АС периферических артерий. Независимым фактором риска развития АС является старение и ассоциированное с ним воспаление стенок сосудов [1]. Одним из ключевых факторов старения является патологическая активация мобильных генетических элементов (МГЭ), таких как ретроэлементы (РЭ) HERV (Human Endogenous Retroviruses) и LINE-1 (Long Interspersed Nuclear Elements-1) [2]. Продукты экспрессии РЭ при старении стимулируют гиперпродукцию интерферона и способствуют вторичным хроническим воспалительным процессам в организме [3]. Для макрофагов характерна экспрессия HERV-K HML-2, коррелирующая с иммунной активацией макрофагов (поляризация в М1-клетки) и ответом на интерферон-I [4]. Согласно новой парадигме иммуностарения, дисфункциональные LB-пенистые макрофаги (CD14+CD16+) продуцируют частицы HERV-K102, высвобождаемые для стимуляции обучаемого врожденного иммунитета [5], что может быть причиной нарушенной экспрессии генов при АС, в том числе активации гена ERVPb1, произошедшего от Env эндогенных РЭ HERV-P [6].
МГЭ – это последовательности ДНК, способные к перемещению в новый локус генома путем «вырезания и вставки» (ДНК-транспозоны) или «копирования и вставки» (РЭ). К автономным РЭ (кодирующим собственные ферменты, необходимые для перемещений) относятся содержащие длинные концевые повторы (LTR) HERV и не содержащие LTR элементы LINE. Неавтономные РЭ используют обратную транскриптазу и эндонуклеазу автономных РЭ для транспозиций, к ним относятся SINE (в том числе Alu) и SVA. РЭ занимают значительную часть генома человека: 8,3% – HERV, 35% – LINE1 и SINE [7]. Роль МГЭ в инициации и развитии АС обусловлена не только опосредованным интерфероном воспалением, но и участием в функционировании иммунной системы. Об этом свидетельствует возникновение необходимых для V(D)J рекомбинации RAG1 и RAG2 от ДНК-транспозонов [8]. Поскольку с возрастом происходит дисбаланс в активации РЭ [2], способствующий старению и воспалению стенок сосудов [1, 3], это может отражаться также на дисрегуляции ДНК-транспозонов и происходящих от них генов V(D)J рекомбинации с последующим дисбалансом иммунной системы при старении [8], что также вероятно отражается на развитии АС (Рис. 1). Активация РЭ при старении может приводить к иммунной патологии также в связи с использованием ERV как энхансеров генов HLA-G и интерферон-индуцибельных генов (формируя транскрипционные сети интерферонового ответа [9]).
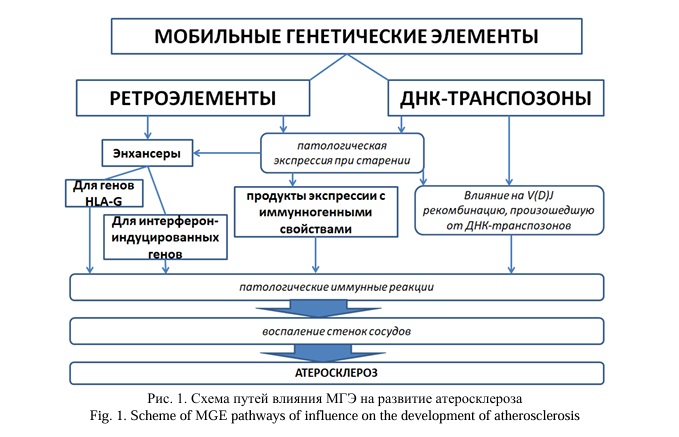
Важную роль в развитии АС играют эпигенетические факторы, изменение которых под влиянием активации РЭ характерно для старения [2]. Под влиянием эпигенетических факторов происходит поляризация ассоциированных с АС макрофагов из противовоспалительных (М2-подобных) в провоспалительные (М1-подобные) и развивается устойчивое воспаление стенок сосудов [10]. Для активированных моноцитов характерна активация HERV-K102 с выходом их продуктов экспрессии в вакуоли, связанные с поверхностями этих клеток, которые превращаются таким образом в «пенистые». Высвобождение HERV-K102 происходит только при лизисе макрофагов. HERV-K102 защищают клетки человека от вирусных инфекций [5], роль которых в развитии АС доказана для вирусов ВИЧ, простого герпеса, гепатита С и В, цитомегаловируса, Т-клеточного лейкоза, папилломы, гриппа [11].
РЭ характеризуются противовирусными свойствами [5] и активацией в ответ на экзогенные вирусы [12], поэтому можно предположить, что одним из механизмов влияния вирусных инфекций на развитие АС может быть изменение экспрессии РЭ. В результате, несмотря на активацию МГЭ в качестве защитного механизма, РЭ могут служить триггерами дальнейшего дисбаланса иммунной системы, в особенности при наличии имеющейся дисрегуляции РЭ, например, при старении [3]. Поскольку МГЭ служат регуляторами экспрессии генов на протяжении всего онтогенеза человека [2, 7], а также драйверами эпигенетических факторов [13], изменение которых обратимо и может быть скорректировано с помощью микроРНК, подробное исследование роли МГЭ и эпигенетических факторов в этиопатогенезе АС определяет возможные новые пути воздействия на эпигенетическую дисрегуляцию с помощью некодирующих РНК (нкРНК), комплементарных МГЭ.
Цель исследования. Определить роль транспозонов и некодирующих РНК в этиопатогенезе атеросклероза и их взаимосвязь между собой в данных процессах.
Материалы и методы исследования. Использованы базы данных Scopus, WoS, PubMed для анализа роли микроРНК, длинных некодирующих РНК, транспозонов в развитии старения и атеросклероза.
Результаты и их обсуждение
Роль некодирующих РНК в патогенезе атеросклероза
К основным эпигенетическим факторам относятся метилирование ДНК, модификации гистонов и РНК-интерференция при помощи нкРНК. При этом нкРНК не только участвуют в посттранскрипционной регуляции экспрессии генов, но и являются ключевыми драйверами модификаций ДНК и гистонов в онтогенезе [13]. Изменения экспрессии микроРНК описаны как патогенетические факторы развивающегося при старении АС [1]. Механизмы участия микроРНК в патогенезе АС связаны с различными механизмами, в том числе с регуляцией метаболизма липидов и воспаления. Эндотелиальное воспаление ассоциировано с повышенными уровнями miR-126, miR-221/222 и низкими уровнями miR10a, miR-155, miR-181a, что ведет к апоптозу, остановке клеточного цикла, выработке активных форм кислорода. При старении эндотелия наблюдается усиление экспрессии miR-217, miR-34; снижение выработки miR-92a, miR-216a, что сопровождается повышением концентраций VCAM (vascular cell adhesion protein), ICAM (intercellular adhesion molecule), MCP1 (monocyte chemoattractant protein 1), CXCL12 (chemokine (C-X-C motif) ligand 12) [14].
Проведенный в 2018 году систематический обзор научной литературы показал, что микроРНК способны контролировать воспаление сосудистой стенки, регулируя ее инфильтрацию активированными лейкоцитами. К ним относятся miR-19a, miR-19b, miR-21. Ключевой микроРНК в данных механизмах АС является miR-126, которая ингибирует VCAM-1 и провоспалительный TNF-α. В связи с этим снижение экспрессии miR-126 повышает активность NF-κB, усиливая взаимодействие лейкоцитов с эндотелиальными клетками (ЭК) и способствуя АС. Влиянием на гладкомышечные клетки сосудов (ГМКС) в патогенезе АС охарактеризованы miR-1 (мишенями являются мРНК генов KLF4, PIM1), miR10a (мРНК гена HDAC4), miR-126 (мРНК генов BCL2, IRS1, FOXO3), miR-22 (ингибирует гены MECP2, HDAC4, EVI1), miR-143 и miR-145 (воздействуют на ACE, ELK1, KLF4/5), miR-21 (мишени – мРНК генов DOCK, PDCD4), miR-26a, miR-34a, miR-130a, miR-221. Воспалительные макрофаги секретируют везикулы, содержащие специфические РНК, липиды и белки, которые используются для коммуникации, в том числе микроРНК между клетками атеросклеротических сосудов (такие как miR-28, miR-146a, miR-185, miR-365, miR-503) [15]. Аномальная пролиферация и миграция ГМКС вовлечены в формирование неоинтимы и способствует рестенозу и образованию бляшек при АС [16].
К эпигенетическим факторам относятся также длинные нкРНК, которые также участвуют в патогенезе АС. Например, длинная нкРНК VINAS [17] влияет на развитие АС за счет регуляции сигнальных путей MAPK и NF-κB, участвующих в воспалении. Нокдаун VINAS снижает экспрессию ключевых воспалительных маркеров, таких как MCP-1, COX-2, TNF-α, IL-1β в ЭК [17]. В плазме крови и в бляшках больных АС определен повышенный уровень длинной нкРНК AK136714, ингибирование которой в экспериментах подавляет формирование АС, воспаление ЭК и защищает эндотелиальный барьер. AK136714 стимулирует транскрипцию Bim, а также напрямую связывается с HuR, повышая стабильность мРНК генов TNF-α, IL-1β и IL-6 [18]. Наблюдаемые изменения уровней длинных нкРНК в патогенезе АС могут быть отражением особенностей экспрессии РЭ, которые служат источниками нкРНК [7]. Это обусловлено высокой чувствительностью РЭ к влиянию средовых воздействий [2, 7, 13], которыми могут служить и экзогенные вирусы.
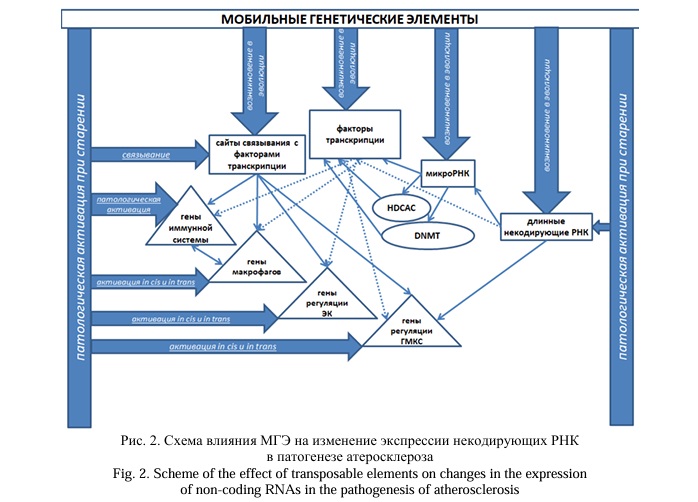
ANRIL взаимодействует непосредственно с последовательностями ретроэлементов Alu в геноме, которые оказывают проатерогенный эффект, располагаясь в областях промоторов генов-мишеней, кодирующих белки группы поликомб PRC-1 и PRC-2. Данные белки рекрутируются с помощью ANRIL и используются для модификации эпигенетических факторов с ингибированием генной экспрессии в цис-регуляции апоптоза, пролиферации и адгезии клеток, воспаления и развития АС [19]. Также выявлено, что модифицированные путем аденозин-инозинового редактирования транскрипты Alu-элементов контролируют стабильность провоспалительной длинной нкРНК NEAT1 при АС. Экспрессия NEAT1, индуцируемая TNF-α, более чем в 2 раза выше в моноцитах крови больных АС коронарных артерий (КА). Подавление NEAT1 приводило к ослаблению вызванной TNF-α провоспалительной реакции эндотелиоцитов, что проявлялось экспрессией CXCL8, CCL2, VCAM1 и ICAM1 [20]. Экспрессия ассоциированной с инфарктом миокарда длинной нкРНК MIAT значительно повышается в сыворотке больных с симптомами нестабильной атеросклеротической бляшки. MIAT действует в качестве губки для miR-149-5p, способствуя экспрессии антифагоцитарной молекулы CD47 [21]. То есть механизм влияния длинных нкРНК на развитие АС может быть связан с регуляцией микроРНК. Данный механизм вероятно обусловлен происхождением в эволюции от МГЭ как длинных нкРНК, так и микроРНК [7, 11, 22] (общее происхождение способствует наличию комплементарных последовательностей). Поэтому логично предположить, что наблюдаемые изменения экспрессии нкРНК при АС являются следствием патологической активации МГЭ при старении [2], которые оказывают не только прямое влияние на развитие АС [1], но и опосредованное, за счет взаимодействий произошедших от них микроРНК и длинных нкРНК (Рис. 2). Доказательство данных механизмов АС может стать основой для таргетного воздействия на патогенез болезни с использованием микроРНК и их миметиков в качестве инструментов, регулирующих патологическую активность МГЭ.
Роль произошедших от мобильных генетических элементов микроРНК в развитии атеросклероза
В базу данных MDTE DB (miRNAs derived from transposable elements database) включены 661 специфических микроРНК, произошедших от МГЭ [22]. Анализ научной литературы позволил нам выявить изменение экспрессии 64 произошедших от МГЭ микроРНК, экспрессия которых изменяется при АС, что связано с различными механизмами их вовлечения в патогенез болезни. У больных АС в экзосомах определены повышенные уровни miR-1202 [23], произошедшей от LTR-ERV1 (полностью соответствует последовательности) [22]. Потенциальной терапевтической мишенью АС может стать miR-1246, возникшая от LTR-ERVL и частично комплементарная его последовательности [22], которая способствует пролиферации, инвазии и дифференцировке ГМКС [24]. Ассоциированная со старением (снижение уровня) [25] miR-1248 подавляет экспрессию тромбомодулина в предшественниках ЭК, что свидетельствует о ее возможном участии в патогенезе АС [26]. MiR-1248 произошла в эволюции от SINE/Alu [22].
У больных АС определено значительное снижение уровня miR-1253, мишенью которой является FOXC2-AS1 (способствует пролиферации ГМКС и ингибирует их апоптоз) [27]. MiR-1253 возникла от LINE2 и от SINE/MIR [22]. MiR-1257, участвующая в путях сборки белков главного комплекса гистосовместимости МНС, регулирует различные гены-мишени, главным образом CALR, а также POMC, TLR4, IL10, ATF6, способствуя прогрессированию АС [28]. Данная микроРНК произошла от ERVL [22]. У пациентов молодого возраста с ишемическим инсультом, обусловленным АС, выявлены повышенные уровни miR-1261 (произошла от ДНК-транспозона Tc-Mar [22]), miR-1290 и miR-891a [29], возникших от SINE/MiR [22]. Низкие уровни miR-1264, сопровождающиеся повышенной экспрессией DNMT1 и фосфорилированного STAT3, определяются при нестабильном АС [30]. У больных инфарктом миокарда в экзосомах, полученных из макрофагов, определены высокие уровни miR-1271 [31], произошедшей от LINE2 [22]. При исследовании образцов коронарных артерий больных АС определено значительное повышение экспрессии miR-1273 [32], семейство которой произошло в эволюции от РЭ LINE, SINE, ERVL [22]. Экспрессия возникшей от SINE/MIR miR-1278 подавляет стимулированную PDGF-BB пролиферацию и миграцию ГМКС при АС [33].
У больных с АС крупных сосудов определено достоверное снижение экспрессии miR-1296 и miR-493 по сравнению с контролем [34]. Данные микроРНК возникли от LINE2. Произошедшая от LINE1 miR-147 [22], обладает атерогенными свойствами, индуцируя экспрессию ICAM-1. С данной микроРНК взаимодействует вовлеченная в АС длинная нкРНК MEG3, действующая как губка для miR-147 [35]. LINE2 является источником miR-151 [22], которая подавляет апоптоз ЭК при АС. Мишенью miR-151 являются IL-17A, а также белок BAX, с-каспазы 3 и 9 [36]. Экспрессия miR-192 (возникла от LINE2 [22]) значительно выше в сыворотке крови больных АС. Данная микроРНК способствует пролиферации и миграции ГМКС [37]. В сыворотке больных АС выявлено значительное снижение уровня miR-211 [38], произошедшей от LINE2. От ДНК-транспозона MER-135 в эволюции возникла miR-224 [22], для которой определена обратная корреляция с АС коронарных артерий у человека [39]. В плазме крови пациентов с нестабильной стенокардией определено значительное повышение уровней miR-28, которая усиливает экспрессию АВСА1, что коррелирует с активацией трансляции мРНК гена LXRα в макрофагах [40]. Данная микроРНК, произошедшая от LINE2 [22], считается потенциальным биомаркером нестабильной стенокардии [40]. В моноцитах периферической крови больных стенокардией выявлено повышение экспрессии miR-2909 (возникшей от LTR-ERVL [22]) при утяжелении окклюзии коронарных артерий с положительной корреляцией. MiR-2909 регулирует гены, вовлеченные в воспаление и иммунитет [41]. Повышенный уровень miR-31 (произошла от LINE2 [22]) вызывает АС за счет воздействия на NOX4 [42]. В плазме крови больных АС определены повышенные уровни miR-3168 [43], возникшей от ДНК-ТЕ hAT Charlie [22].
У больных АС коронарных артерий значительно повышена экспрессия miR-320b, которая регулирует отток холестерина из макрофагов. Введение miR-320b экспериментальным животным увеличивало размеры атеросклеротических бляшек, содержание поврежденных макрофагов и уровни провоспалительных цитокинов за счет усиления фосфорилирования NF-κB [44]. Источником miR-320b в эволюции является LINE2 [22]. Произошедшая от LINE2 miR-325 [22] способствует развитию АС за счет подавления экспрессии KDM1A, снижая уровни SREBF1 и ингибируя активацию пути PPARγ-LXR-ABCA1 [45]. В образовании окисленных пенистых клеток при АС определена роль miR-326 (произошла от ДНК-транспозона hAT-Tip100 [22]), вовлеченная в сеть взаимодействий кольцевых РНК с длинными нкРНК [46]. Концентрация возникшей от SINE/MIR miR-335 [22], повышена в плазме крови больных АС [43]. В макрофагах, ГМКС и ЭК при атерогенезе определяется повышение экспрессии miR-340 [47], произошедшей от ДНК-транспозона TcMar-Mariner [22]. В периферических мононуклеарах определены высокие уровни miR-342 [48] (возникшей от SINE/tRNA-RTE [22]), которые положительно коррелировали с концентрациями в сыворотке крови IL-6 и TNF-α [47].
В сыворотке больных АС определено значительное повышение экспрессии miR-3646 [49] (произошедшей от SINE/MIR [22]) и miR-374 (произошла от LINE2 [22]), которая стимулирует пролиферацию и миграцию ГМКС [50]. Снижение оттока свободного холестерина из макрофагов и усиленный приток окисленных липопротеинов низкой плотности является важным фактором развития АС. В метаболических путях, регулирующих эти процессы, участвует произошедшая от SINE/MIR и LINE2 [22] miR-378 [51]. Ускоряет развитие АС за счет влияния на макрофаги (нарушая их аутофагию) также miR-384 [52], произошедшая от LINE-Dong-R4 [22]. Низкая экспрессия miR-421 (произошла от LINE2 [22]) в сыворотке, бляшках и ГМКС у больных АС коронарных артерий повышает уровни CXCL2 [53]. При АС определяется также снижение концентрации miR-4286, возникшей от ERVL [22] и ингибирующей TGF-β1 (способствует повреждению ЭК) [54]. Возникшая от ДНК-транспозонов hAT Charlie miR-4463 [22], препятствует переключению фенотипа ГМКС, способствуя АС [55]. MiR-4487 (произошла от LINE1 [22]) при АС стимулирует миграцию и выживаемость ГМКС и ингбирует их апоптоз путем целевого воздействия на RASA1 [56]. MiR-4731 (источник – LINE-CR1 [22]) вызывает пролиферацию и миграцию ГМКС, взаимодействуя с транскрипционным фактором FOXO3 и длинной нкРНК SENCR (которая обладает противоположным эффектом) [57].
MiR-487 (произошедшая от SINE/MIR [22]) предложена в качестве молекулярной мишени для лечения АС. Данная микроРНК ингибирует р53 и СВР, усиливая пролиферацию ЭК [58]. MiR-495 (источник – ERVL [22]) участвует в патогенезе АС путем связывания с кольцевой РНК hsa_circ_0126672 [59]. MiR-498 (произошла от LINE1 [22]) оказывает посттранскрипционное ингибирование на ген SCD (stearoyl-CoA desaturase), который в норме снижает уровень холестерина в сыворотке. У людей с полиморфизмом rs41290540CC в 3’UTR этого гена, нарушающий связывание с miR-498, определен низкий риск АС КА [60]. Уровень произошедшей от LINE2 miR-502 [22] значительно повышен в сыворотке больных АС коронарных артерий [61]. MiR-511 (источник – LINE1 [22]) является одним из «связующих» компонентов мультисубъединичного комплекса, участвующего в терминальных стадиях синтеза холестерина с регуляцией семейства белков GPCR, которые вовлечены в трансформацию патологических фенотипов ГМКС при АС [62]. MiR-520d (произошла от SINE/Alu [22]) ингибирует экспрессию гена PCSK9, вызывающего деградацию рецепторов липопротеинов низкой плотности, подавляя развитие АС [63].
MiR-544 (источник в эволюции – hAT Charlie [22]) участвует в патогенезе АС за счет регуляции формирования и репарации ЭК, способствуя созреванию и антиоксидантным свойствам ЭК путем регулирования сигнальных путей YY1/TET2 [64]. У пациентов с АС в жировой ткани вокруг коронарных артерий определено снижение экспрессии miR-548. Представители семейства данной микроРНК произошли в эволюции от различных РЭ (LINE1, LINE2, LTR-ERVL, LTR-Gypsy, LTR-ERV1, SINE/MIR) и ДНК-транспозонов (TcMar, hAT Charlie) [22]. MiR-548 регулирует экспрессию гена HMGB1 (кодирует негистоновый белок, связывающий хроматин и участвующий в контроле транскрипции, репликации и репарации ДНК) [65]. Повышенная экспрессия miR-552 (произошла от LINE1 [22]) под влиянием PDGF-bb определена в ГМКС, что ведет к стимуляции их пролиферации, инвазии и миграции. Мишенями miR-552 являются мРНК генов SKI и ATF4 [66]. MiR-575, произошедшая от ДНК-транспозона hAT Charlie [22], предложена в качестве биомаркера и клинической мишени у больных АС. Данная микроРНК ингибирует миграцию и пролиферацию ЭК и стимулирует их апоптоз. MiR-575 подавляет экспрессию мРНК гена Rab5B [67]. Кольцевая РНК circ_0086296 индуцирует АС через петлю обратной связи IFIT1/STAT1, действуя как губка для miR-576 (возникла от LINE1 [22]), которая ингибирует экспрессию IFIT1-STAT1, препятствуя развитию АС [68]. Hsa_circ_0031891 подавляет miR-579, усиливая экспрессию HMGB1 и PDGF-BB-индуцированную пролиферацию, миграцию и нарушение дифференцировки ГМКС аорты человека. Экспрессия miR-579 снижена у больных АС коронарных артерий [69]. Произошедшая от LINE-CR1 [22] miR-582 определяется на высоком уровне в сыворотке больных АС [43].
При воспалительных реакциях снижается уровень эндотелиальной синтетазы оксида азота (eNOS), которая является главным регулятором гомеостаза ЭК. При АС отсутствие OASL1 (2’-5’ oligoadenylate synthetase-like 1), необходимой для поддержания стабильности мРНК eNOS ускоряет прогрессирование бляшек. OASL1 взаимодействует с miR-584 (произошедшей от ДНК-транспозона hAT-Blackjack [22]), которая ингибирует мРНК eNOS, связываясь с ее 3’UTR [70]. Сверхэкспрессия miR-612 (произошла от SINE/MIR [22]) ингибирует миграцию и инвазию ГМКС, вызывая остановку клеточного цикла на стадии G1. Под влиянием PDGF-BB, который способствует пролиферации и миграции ГМКС, снижается экспрессия miR-612 [16]. В сыворотке больных АС снижен уровень PON1 и длинной нкРНК, действующей как конкурентная эндогенная РНК для miR-616 (произошла от LINE2 [22]). Было определено, что miR-616 ингибирует экспрессию PON1, способствуя развитию АС [71].
Кольцевая РНК circARHGAP12 в экспериментах на мышах способствовала АС за счет стимуляции пролиферации и миграции ГМКС аорты. CircARHGAP12 также напрямую связывалась с miR-630 (возникла от SINE/MIR [22]), мишенью которой является метилтрансфераза гистонов EZH2, модулирующая транскрипцию TIMP2 в регуляции миграции ГМКС и вызывающая развитие АС [72]. Сходным механизмом действия обладает hsa_circ_0008896, влияющая на ГМКС посредством взаимодействия с miR-633 (произошедшая от SINE/MIR [22] и регулирующая CDC20B) [73]. MiR-637 (источник – LINE1 [22]) ингибирует экспрессию TRAF6 и способствует пролиферации ЭК и ангиогенезу, ингибируя апоптоз и воспаление. Взаимодействующая с ней circ_0003575 вызывает обратный эффект, а также активирует путь NF-κB [74]. Экспрессия miR-641 (произошла от SINE/MIR [22]) снижена в индуцированных окисленными липопротеинами низкой плотности ГМКС. С данной микроРНК взаимодействует длинная нкРНК MIAT [75]. Источником miR-652 в эволюции является ДНК-транспозон hAT-Tip100 [22]. Ингибирование этой микроРНК уменьшает прогрессирование АС и усиливает восстановление эндотелия за счет стимуляции экспрессии циклина D2 [76].
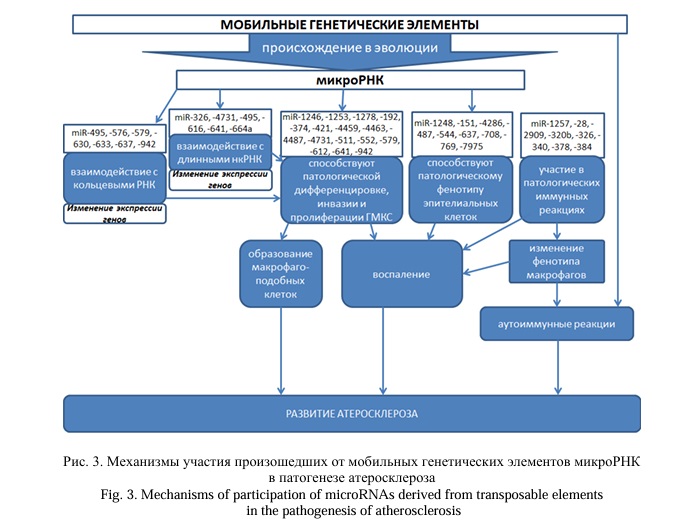
У больных АС определена пониженная экспрессия miR-664a (произошла от LINE1 [22]) [77]. Длинная нкРНК Punisher регулирует апоптоз и митохондриальный гомеостаз ГМКС посредством взаимодействия с miR-664a [77]. Произошедшая от LINE2 miR-708 [22] экспрессируется на высоком уровне в ЭК неоинтимы в поврежденных сосудах при физиологическом потоке крови и не экспрессируются при застое. MiR-708 обладает противовоспалительным свойством, подавляя экспрессию связанной с рецептором интерлейкина-1 киназы, рецептора интерлейкина-6, консервативной сприаль-петля-спираль вездесущей киназы и ингибитора субъединицы-γ киназы ядерного фактора κB [78]. В тканях артерий у больных АС определены повышенные уровни произошедшей от LINE/CR1 [22] miR-769, мишенью которой являются мРНК генов киназы GSK3B и TRAPPC2B [43]. MiR-7975, произошедшая от LTR-ERV1 [22], предложена в качестве потенциального биомаркера и мишени для лечения АС [79]. Уровни экспрессии miR-942 (произошедшей от LINE2 [22]) оказались достоверно снижены у больных в постбифуркационных каротидных АС. miR-942 подавляет экспрессию гена семейства адгезинов GPR56 [80]. Таким образом, произошедшие от МГЭ микроРНК могут влиять на развитие АС посредством изменения экспрессии генов в ГМКС (способствуя патологическиой пролиферации, дифференцировке, инвазии и апоптозу клеток), в ЭК (вызывая патологическую экспрессию генов в клетках) и макрофагах, а также влияя на иммунные процессы (miR-1257 [28]; miR-28 [40]; miR-2909 [41]), эпигенетические факторы (miR-1264 [30], miR-630 [72], взаимодействуя с длинными нкРНК [46, 57, 71, 75, 77] и кольцевыми РНК [59, 68, 69, 72, 73, 80] (Рис. 3).
Роль ассоциированных с атеросклерозом произошедших от мобильных генетических элементов микроРНК в старении
Поскольку АС связан со старением и воспалением стенок сосудов [1], активацией МГЭ [2, 3] при старении, проведен анализ научной литературы об ассоциации произошедших от МГЭ микроРНК одновременно с АС и старением. В фибробластах человека при старении определено повышение уровней miR-1246, miR-1271 [81, 82], miR-1273 [81], miR-1290 [82], а также снижение уровней miR-1257 [81], miR-1261 [82]. При старении определено снижение экспрессии miR-1248, miR-151 [25] и miR-147 [83], повышение экспрессии miR-192 [84]. Уровень miR-211 значительно выше у долгожителей по сравнению с людьми с короткой продолжительностью жизни (низкая экспрессия), обратная корреляция определена для miR-340 и miR-374 [85].
MiR-224 ассцоиирована со старением головного мозга. Ее мишенью является мРНК гена CHOP, вовлеченного в регуляцию митохондриальных белков [86]. В ранних стадиях старения ЭК определено транзиторное снижение концентрации miR-28 в данных клетках [87]. MiR-31 действует как ключевой драйвер старения фолликулярных стволовых клеток волос путем прямого нацеливания на мРНК гена Clock (основной ген циркадных часов, нарушение регуляции которого активирует каскад MAPK/ERK), вызывая истощение HFSC посредством трансэпидермальной элиминации. Условная абляция miR-31 обеспечивает эффективную защиту кожи от старения [88]. Повышенная экспрессия miR-320b ассоциирована со старением [89]. Снижение уровней miR-325 способствует старению хондроцитов за счет активации путей p53/p21 [90]. Повышенная экспрессия miR-326 определяется в фибробластах кожи при старении [91].
Повышенная экспрессия miR-335 способствует старению ЭК, ингибируя экспрессию гена sKlotho [92]. При старении в мононуклеарах периферической крови определено снижение экспрессии miR-342, нацеленной на мРНК гена SIRT6 [93]. Повышение экспрессии miR-378 определено у людей старческого возраста при регенерации мышц. Мишенями miR-378 являются мРНК генов сигнальных путей инсулино-подобного фактора роста (IGF-1) [94]. Было выявлено, что miR-384 негативно регулирует возрастную остеогенную дифференцировку мезенхимальных стволовых клеток костного мозга, что свидетельствует о роли данной микроРНК в старении [95]. Со старением кожи определена ассоциация повышения экспрессии miR-4487 [96].
В экспериментах на клеточных линиях человека miR-495 способствовала старению мезенхимальных стволовых клеток за счет воздействия на мРНК протоонкогена BMI1 [97]. MiR-520d способствует старению скелетной мускулатуры за счет влияния на регуляторные факторы MyoD, MyoG, Mef2c, Myf5. Длинная нкРНК GPRC5D-AS1, ингибирующая miR-520d, предложена в качестве терапевтической мишени для лечения саркопении [98]. При старении у людей определено увеличение экспрессии miR-552 в 124 раза большее в сравнении с молодыми людьми [99]. Со старением фибробластов человека ассоциировано снижение уровней miR-548, miR-576 и повышение – miR-584 [81, 82], miR-633 [83], miR-641 [81]. В экспериментах при старении суставов было определено снижение уровней miR-652 и miR-708 [100]. В таблице 1 представлены данные об изменениях экспрессии специфических микроРНК, произошедших от МГЭ при атеросклерозе и об их ассоциации со старением.
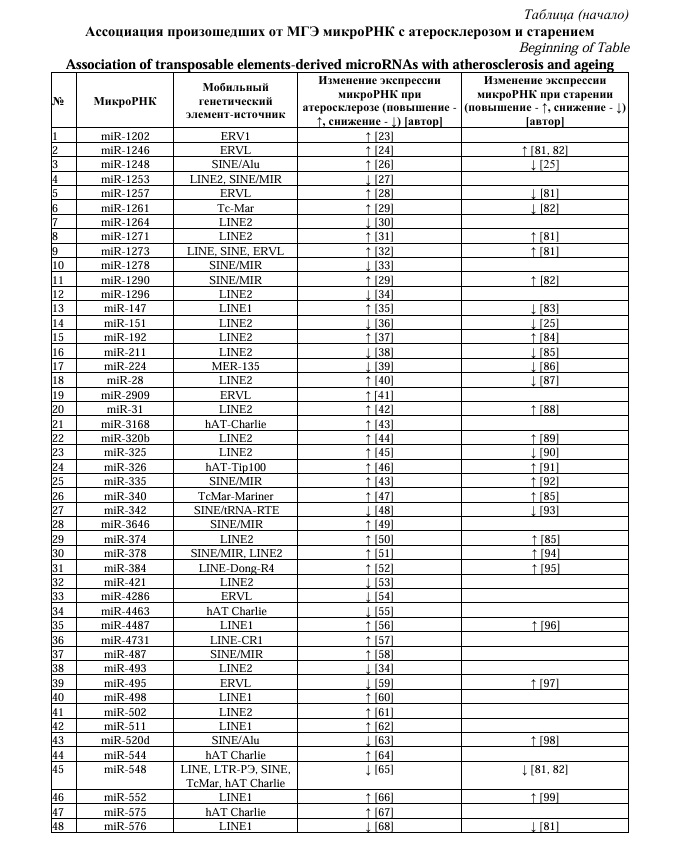
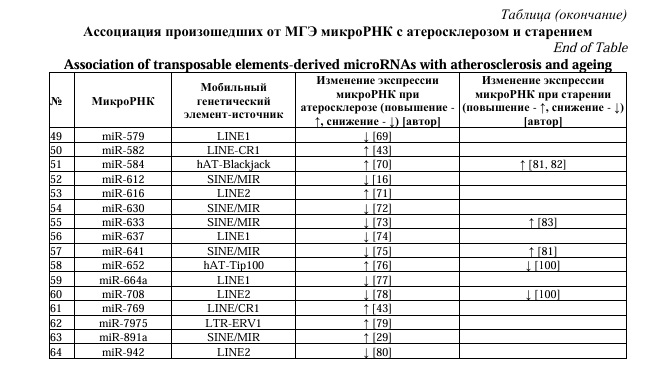
Представленные в таблице данные свидетельствуют о том, что активация МГЭ при старении может служить инициирующим событием в нарушении регуляции эпигенетических генных сетей, что отражается в изменениях уровней специфических микроРНК, способствуя атеросклерозу. При АС может быть сходное со старением возрастание уровней произошедших от МГЭ микроРНК (miR-1246, -1271, -1273, -192, -31, -320b, -326, -335, -340, -374, -378, -384, -4487, -552, -584) или их снижение (miR-151, -211, -224, -342, -421, -493, -548, -576, -708). Для нескольких микроРНК определено разное изменение экспрессии при старении и атеросклерозе (miR-1248, -1257, -1261, -1290, -147, -28, -325, -495, -520d, -633, -641, -652), что логично, поскольку старение является не единственным фактором, способствующим развитию болезни. Активация МГЭ также происходит под влиянием различных причин, в том числе вирусов [11] и стресса [7], как было отмечено в статье.
Заключение. Увеличение заболеваемости атеросклерозом с возрастом можно объяснить ролью в старении активации МГЭ, которые вызывают патологическую активацию микроРНК и длинных нкРНК, генов иммунной системы, макрофагов, ЭК, ГМКС. Кроме того, продукты экспрессии РЭ являются триггерами интерферонового ответа и развития асептического воспаления в организме, характерного для АС. Анализ научной литературы позволил выявить 64 произошедших от МГЭ микроРНК (30 – от LINE, 13 – от SINE, 10 – от ДНК-транспозонов, 7 – от LTR-содержащих РЭ, 2 – одновременно от LINE и SINE, 1 – от LINE, SINE, LTR, 1 – от РЭ и ДНК-транспозонов), изменения экспрессии которых ассоциированы с атеросклерозом. Механизм их участия в патогенезе АС обусловлен влиянием на экспрессию генов в эндотелиоцитах, в гладкомышечных клетках сосудистой стенки, в макрофагах; воздействием на метаболизм липопротеинов; изменением функционирования длинных нкРНК и кольцевых РНК. Из 64 ассоциированных с атеросклерозом произошедших от МГЭ микроРНК при старении определено изменение экспрессии 34 микроРНК. Это свидетельствует о наличии общих эпигенетических механизмов старения и атеросклероза, обусловленных патологической активацией МГЭ.
Информация о финансировании
Финансирование данной работы не проводилось.














Список литературы