
In-silico insights into α-Synuclein mediated pathology of Parkinson’s disease using molecular docking and molecular dynamics simulation studies
Aннотация
Background: The precise cause of neuronal loss in Parkinson's disease (PD) is multifactorial, involving genetic and environmental factors. Genetic mutations, affecting many proteins such as α-Synuclein, Leucine-rich repeat kinase 2, and Parkin, have been implicated in sporadic and familial cases, shedding light on key molecular pathways involved in disease pathogenesis. The aim of the study:The present in-silico study was undertaken to study the hub protein responsible for PD pathogenesis and the effect of mutations on its pathophysiology. Materials and methods: Pathological proteins were selected using the KEGG database, and their protein-protein interaction mapping was done using the STRING database, followed by network merging and hub-protein identification using Cytoscape 3.9.1. Protein-protein docking studies were performed to study the pathology of identified hub-protein using Hex 8.00, and their validation was done by performing molecular dynamics simulation studies using GROMACS v2020.1. Results: α-Synuclein was identified as the hub protein responsible for PD pathology and studied for its pathological mechanism of aggregation in wild-type and mutated form (A53T, E46K, H50Q, A53E and G51D) using protein-protein docking studies. On decamerization, 4 of 5 studied SNPs (A53T, A53E, G51D and H50Q) showed better binding affinities in the VI-IV combination, while E46K showed better binding affinity in the VII-III combination than wild-type α-Synuclein. The SNPs – A53T, E46K and H50Q, demonstrated lower binding energies while 2 SNPs (A53E and G51D) displayed higher binding energies in decameric form than wild-type (WT) α-Synuclein aggregates. Also, G51D and E46K mutated oligomeric structures of α-Synuclein showed twisted morphologies. Molecular dynamics simulation studies provided evidence for the stabilized conformation of the decameric form of wild-type α-Synuclein. Conclusion: The study paves a good platform for further investigation to consider the decameric form of α-Synuclein protein as a target for PD therapeutics
Ключевые слова: Parkinson’s disease, hub-protein, α-Synuclein, single nucleotide polymorphisms, aggregation, oligomerization, protein-protein docking, molecular dynamics simulation
К сожалению, текст статьи доступен только на Английском
Introduction. Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by the selective degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNc) of the brain [1]. The loss of dopaminergic neurons in the SNc leads to a decrease in dopamine levels in the striatum, thus playing a critical role in motor control and imbalances in the activity of striatal spiny projection neurons (SPNs) [2]. In the direct pathway of basal ganglia, the activity of striatal SPNs is reduced, leading to decreased facilitation of movement. Conversely, in the indirect pathway, the activity of striatal SPNs is increased, resulting in excessive inhibition of movement [3, 4]. These imbalances in the direct and indirect pathways contribute to the motor symptoms observed in PD, leading to bradykinesia, rigidity, and tremors.
The pathogenesis of PD involves a complex interplay between genetic and environmental factors. Genetic risk factors have been implicated in the development of sporadic and familial PD. Mutations in the SNCA gene, encoding the α-Synuclein protein, are found in familial forms of PD and are also associated with sporadic cases [5, 6]. Other genes associated with familial PD include Parkin [7], DJ-1 [8], PINK1 [9], and LRRK2 [10]. These genetic mutations disrupt various cellular processes within neurons and contribute to the neurodegenerative process in PD.
Understanding the underlying mechanisms of PD, the main protein targets, and the contributions of genetic mutations are crucial elements for the development of effective therapeutic strategies. Research efforts focused on elucidating these mechanisms are essential for identifying targets for disease-modifying interventions and improving the therapeutic potentials of PD. The present study was carried out to identify the major therapeutic target protein(s) involved in the cause and progression of PD. Also, the study attempted to have in-silico insights into genetic variation(s), if any, of these target protein(s) and their role in PD.
Materials and Methods
Identification of target proteins and their protein-protein interactions
Different proteins/ protein-coding genes involved in the pathophysiology of Parkinson’s disease were identified using the KEGG pathway [11] database for diseases, with KEGG ID: map05012.
The protein-protein interaction (PPI) networks of these identified proteins/ protein-coding genes were retrieved using the STRING [12] database and a single merged protein-protein interaction network map was generated with a confidence score of 0.40, using Cytoscape 3.9.1 [13].
Hub-protein identification
The top 10 hub proteins involved in PD, were identified by selecting the “merged network” as the target network and the node score was calculated. The Maximal Clique Centrality (MCC) topological algorithm of the CytoHubba module [14] was used for Hub-proteins’ score calculation. The Analyzer tool of Cytoscape 3.9.1 was used to analyse the merged network.
Retrieval and energy optimization of the tertiary structure of identified hub-protein
The tertiary structure of the identified hub protein, involved in the pathophysiology of PD was retrieved from the RCSB Protein Data Bank (PDB) [15] and was subjected to energy minimization using UCSF Chimera [16] using default parameters, i.e., 100 steps of steepest descent (step size of 0.02 Angstrom) and 10 steps of the conjugate gradient algorithm (step size of 0.02 Angstrom), taking AMBERff14SB as the force field parameter, along with the addition of Gasteiger charges. The energy-minimized structure was saved in the PDB file format for further studies.
Text mining for genetic mutations in identified hub-protein and their structure modelling
An extensive literature search was performed to identify the non-synonymous SNPs/point mutations in the identified hub protein, reported to affect the pathophysiology of PD. The 3D structures of the mutated hub protein reported earlier were constructed by homology modelling using the Swiss Model [17]. The modelled structures were subjected to energy minimization and optimization using UCSF Chimera [16], using default parameters as mentioned in Section 2.3.
Protein-protein docking study
Protein-protein docking was carried out to study the role of mutations in Parkinson’s disease, using Hex 8.0.0 with default parameters i.e. Correlation type – Shape + Electro; FFT Mode – 3D; Sampling method – Range angles; Grid dimension – 0.6; Solutions – 2000; Receptor range – 180; Step size – 7.5; Ligand range – 180; Step size – 7.5; Twist range – 360; Step size – 5.5; Distance range – 40; Box size – 10; Steric scan – 18; Final search – 25 [18].
Molecular dynamics simulation study
The stability of the protein-protein docked complex of the decameric form of the identified hub protein was analyzed by performing molecular dynamics simulation [MDS] using GROMACS v2020.1 [19]. The topology file of the protein was generated using charmm36m [20] force field. Solvation of the system was done by adding TIP3P water molecules followed by neutralization using Sodium (Na+) and Chloride (Cl-) ions at a concentration of 0.1M. For maximum stability, energy minimization of the system was performed using the steepest descent algorithm (5,000 steps) and potential energy of the system was lowered up to 1,000kJ/mol. Equilibration of the system was performed under constant pressure (1 bar) and temperature (310K) conditions for 10ns using the Parrinello-Rahman barostat and Nose-Hoover thermostat, respectively. MDS was performed for 100ns using periodic boundary conditions and trajectories were recorded for every 100ps. Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), Radius of gyration (Rg) and Solvent Accessible Surface Area (SASA) were calculated for each frame during MDS analysis.
Results and discussion
Identification of target proteins and their protein-protein interaction study
A total of 7 protein-coding genes, majorly involved in the pathophysiology of PD, were identified using the direct and indirect pathways, as retrieved from the KEGG database. Both the pathways had 7 common target proteins i.e., α-Synuclein (SNCA), Parkin, Ubiquitin carboxyl-terminal hydrolase L1 (UCHL1), Leucine-rich repeat kinase 2 (LRRK2), PTEN -induced kinase 1 (PINK1), Deglycase (DJ1) and High-temperature requirement A (HTRA2).
Retrieval of protein-protein interactions
The protein-protein interaction (PPI) networks of the identified target proteins were retrieved from the STRING database (Fig. 1a-g). These networks were analysed for the number of nodes, edges, and average local clustering coefficient. The PPI networks of all the studied proteins displayed 11 nodes (depicting proteins) with the number of edges (depicting protein-protein associations) varied from 24 to 43 while the average clustering coefficient was observed to be around 0.8 (Table 1). Edges displayed two types of interactions, known interactions which are either experimentally determined or retrieved from curated databases; and predicted interactions based on gene fusion, gene neighborhood, and gene co-occurrence. The merging of individual protein-protein interaction networks of all the studied proteins resulted in a single merged network map, which was used for identifying hub protein in the pathophysiology of PD.
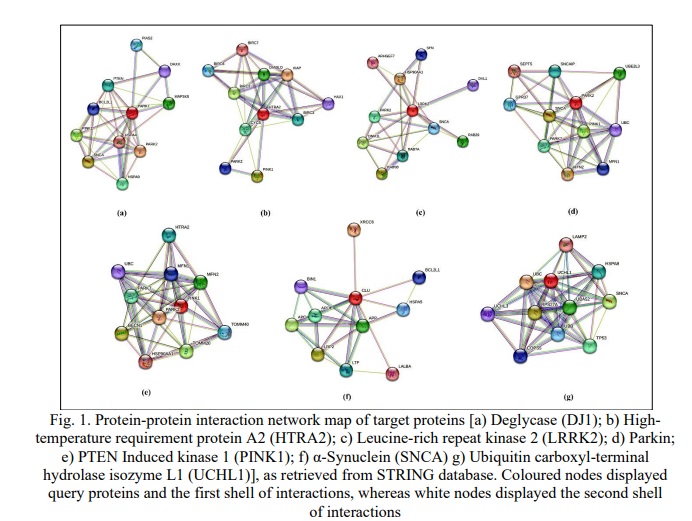
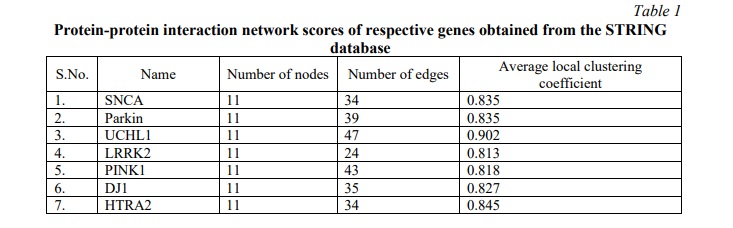
Hub-protein identification
The top 10 hub proteins were identified from the merged network map, based on the neighbourhood scores of nodes and edges using the MCC algorithm of the CytoHubba module (Table 2; Fig 2). The Analyzer tool provided the summary statistics of the undirected network map of the top 10 hub proteins (Table 2, 3) and the average number of neighbors was observed to be 7.600 with a clustering coefficient of 0.927. α-Synuclein (SNCA) was identified as the top-ranked protein, having the highest MCC score of 12418, whereas UBC (Polyubiquitin-C) and UBB (Polyubiquitin-B) with respective MCC scores of 12408 and 11500, were identified as the second and third proteins of the top 3 scoring nodes.
α-Synuclein is known to exist in dynamic equilibrium between monomeric to oligomeric forms and vice-versa [21]. Monomeric α-Synuclein may combine to form a pathological conformer that follows a reversible pattern of aggregation, leading to the formation of oligomers, protofibrils and amyloid fibrils respectively. Multiple stacks of amyloid fibrils form irreversible circular disease-causing aggregations called Lewy bodies [22]. These aggregates are resistant to degradation due to the impaired ubiquitination system (UBB and UBC) [23]. α-Synuclein being the primary component of Lewy bodies, was selected for further studies of PD pathology.
Genetic mutations in identified hub-protein
Monomeric α-Synuclein (140 amino acids) has a differentially marked amphipathic amino-terminus (residue 1-60) – the site of single nucleotide polymorphisms (SNPs); hydrophobic non-amyloid-β component (NAC) domain (residue 61-95) – that forms the aggregation core; and acidic carboxy-terminal (residue 96-140) – the site of metal ion binding [21]. Various point-mutations or SNPs- A53T (dbSNP ID: rs104893877), A53E (dbSNP ID: rs1553408288), E46K (dbSNP ID; rs104893875), H50Q (dbSNP ID: rs201106962) and G51D (dbSNP ID: rs431905511), in the amphipathic region of α-Synuclein, are reported to play possible roles in its rate of self-aggregation [24-27]. Hence, the role of these SNPs of α-Synuclein, on its self-aggregations, was analyzed using the in-silico tool of protein-protein docking.
Tertiary structure retrieval and optimisation of the identified hub-protein
The common kernel structure with preNAC and NAC regions of α-Synuclein (PDB ID: 6CU8), reported to show better self-association/aggregation properties [28], was selected for our study of self-aggregation rate and patterns of mutated α-Synuclein. The selected conformation had 10 chains of α-synuclein, so all chains except chain-A, were deleted and its energy-minimized structure was used for further docking studies.
Homology modelling and structure optimization of mutated hub-protein
Individual mutated models of selected five SNPs of α-Synuclein (A53T, A53E, E46K, H50Q, and G51D) were constructed by homology modelling using the Swiss Model, in template mode with PDB ID: 6CU8, selected as template. The modelled structures of mutated α-Synuclein were subjected to energy minimization and root mean square deviation (RMSD) was calculated using Chimera. A significant RMSD was observed between monomeric units of mutated α-Synuclein with respect to wild-type (WT) α-Synuclein, ranging from 0.108 to 0.118 (Table 4).
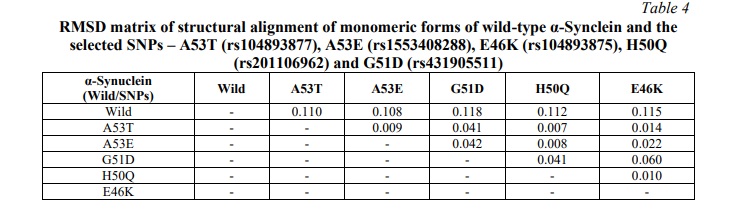
Protein-protein docking to study the effect of mutations on multimerization
Protein-protein docking studies were carried out successively, with dimer-monomer docking to form a trimer, followed by dimer-dimer and trimer-monomer docking to form a tetramer and so on till decamerization, to evaluate the differential pattern of aggregations of WT and mutated forms of α-Synuclein proteins and to analyse the effect of SNPs or point-mutations on aggregation/self-assembly of α-Synuclein proteins.
The analyses of the docking studies (Tables 5a-c) revealed that for tetramerization, we had two possible combinations of trimer-monomer (III-I) and dimer-dimer (II-II). All the mutated and the WT structures showed better affinity to self-associate in the II-II combination except for the SNP E46K, which showed better results in the III-I combination (Table 5a). For the Pentamer, we had two combinations, III-II and IV-I. All the models showed better affinities in the IV-I combination. For Hexamer, a total of three combinations V-I, IV-II and III-III were evaluated for oligomerization. Except for the A53T, which showed lower energy in the IV-II combination, all others showed lower energies in the V-I combination hence, the better affinity than other studied combinations (Table 5a).
The heptamerization also had 3 possible combinations, V-II, IV-III and VI-I. The WT and SNPs- E46K and H50Q, had better affinities in VI-I pairing, while A53E and G51D had lower energies in the V-II combination and A53T showed better affinity for IV-III combination (Table 5b). Four possible octamerisation combinations were investigated in our study: V-III, VI-II, VII-I and IV-IV. All the studied models of SNPs showed better affinity to self-associate in the IV-IV combination except for E46K, which had lower energy in the VII-I combination and showed a twisted morphology in the IV-IV pairing (Table 5b).
For nonamerization, again we had four combinations, V-IV, VI-III, VII-II and VI-III. WT, A53T, G51D, and H50Q had lower energies in V-IV combinations, while A53E and E46K showed better affinities in VII-II and VI-III combinations respectively (Table 5c). In the case of Decamerization, we had five possible combinations, VIII-II, V-V, VI-IV, VII-III and IX-I for evaluation. Except for the E46K, all other models gave better affinities in the VI-IV combination. However, E46K showed better affinity in the VII-III combination (Table 5c).
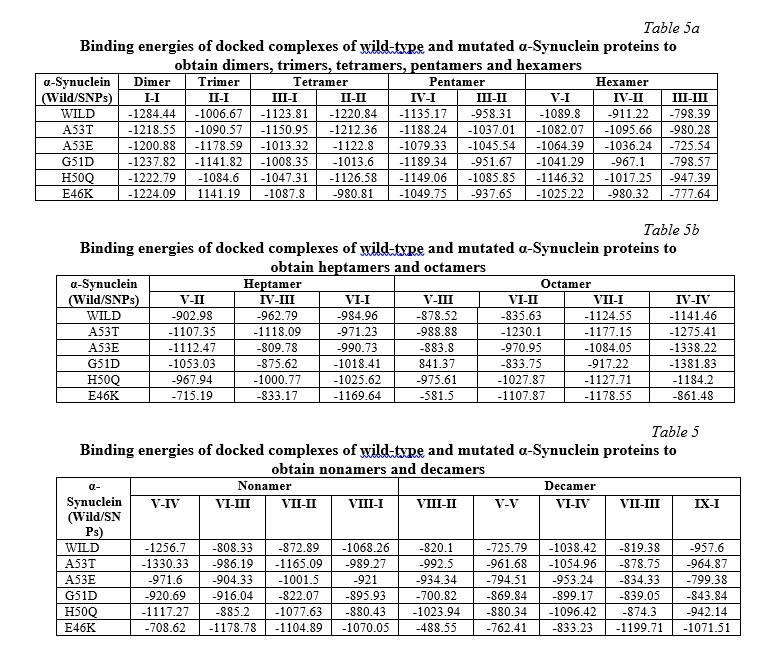
Analysis of the effect of mutations on aggregation/self-assembly
Our study revealed altered binding energies (Table 6) and morphologies (Fig. 3a-f) of the mutated oligomeric structures in comparison to WT oligomeric structures of α-Synuclein. On self-assembly of A53T mutant, a lower binding energy (-1054.96 KJ/mol) was observed at the decamer stage i.e. oligomeric form, in comparison to WT α-Synuclein (-1038.4 KJ/mol), indicating high affinity of oligomerization of the former structure. A low affinity of self-assembly was observed for the A53E mutant, having higher binding energy (-953.24 KJ/mol) on decamerization. The findings were observed to be in good concurrence with earlier reports [29, 30], which showed that the substitution of alanine with threonine at the 53 position in the A53T mutant formed a steric zipper in the core region of α-Synuclein oligomers, explaining the tendency of A53T mutant to aggregate at a higher rate compared to WT α-Synuclein while the substitution of Alanine with Glutamate at 53 position (A53E) has been reported to decrease the rate of aggregation/ self-assembly [29].
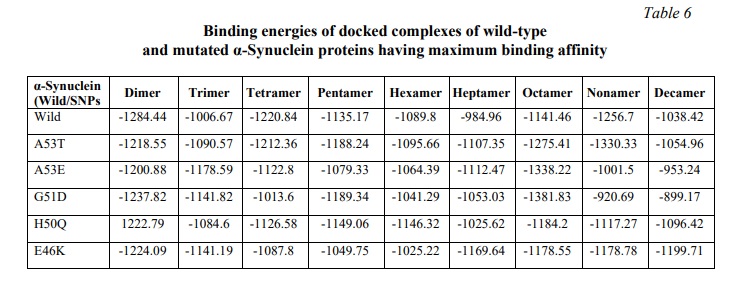
For the E46K mutation, lower binding energy (-1199.71 KJ/mol) was observed in comparison to WT (-1038.4 KJ/mol) and A53T (-1054.96 KJ/mol) mutant of α-Synuclein, along with a twisted morphology at the nonamer and decamer stages (Fig. 3d). This observation can be inferred from its high affinity of self-aggregation as compared to WT and A53T mutant of α-Synuclein concurrent to the previous studies [29, 30], which explained the formation of stabilizing salt bridge due to the close proximity of E46 and K80, causing enhanced electrostatic repulsion and increased propensity of oligomerization.
The present docking study of H50Q mutation of α-Synuclein for self-aggregation revealed a lower binding energy (-1096.42 KJ/mol) as compared to WT α-Synuclein (-1038.4 KJ/mol). The observation was also in accordance with the earlier reports [26, 30], which explained that the substitution of His with Gln eliminated the imidazole's positive charge, thereby, preventing the formation of a salt bridge with adjacent filament. Just like E46K, in the case of H50Q also, there is a destabilization of the interface between the protofibril dimers, creating a potential shift in the equilibrium favoring self-aggregation in α-Synuclein.
For the G51D mutant, a significant rise in the binding energy (-899.17 KJ/mol) was observed in higher oligomeric units like nonamer and decamer, in addition to the disrupted morphology (Fig. 3e). The reduced binding affinity of the G51D mutant in later stages of the oligomerization may be inferred as its decreased rate of aggregation, in concurrence with the previous studies [26, 30], which elucidated that the additional steric bulk was responsible for the loss of flexibility and hydrophobicity of G51D maturated α-Synuclein, causing the decreased rate of self-assembly.
Molecular dynamics simulation study
The stability of the decameric form of WT α-Synuclein was studied by molecular dynamics simulation (MDS) and observed to be stable throughout the period of 100 ns. The root mean square deviation (RMSD) value was found to increase gradually from a time scale of 0.1ns to 36.7ns, and after that, the RMSD value is observed to be stable within the 3Å (0.30 nm) limit, varying within the lowest of 0.61 at 36.8ns and a highest of 0.85 at 69 ns up to 100ns (Fig. 4a). Thus, validating the docked complex of the WT α-Synuclein.
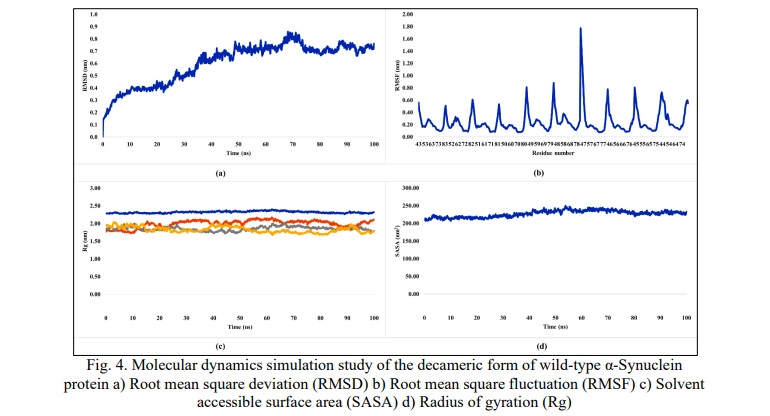
The root mean square fluctuation (RMSF) was calculated (Fig. 4b), and major peaks were observed for the tails (N and C-terminal) and minor peaks were observed for the loop region of each monomeric unit (43-83 amino acids). The beta-sheet domain was observed to be stable for each monomeric unit in the decameric conformation, indicating a stable docked complex of WT α-Synuclein.
The solvent-accessible surface area (SASA) was observed to be constantly varying between 200-250nm2. This small change in SASA explains the minor changes in the structure of the docked complex along the simulation and hence reflects towards stability of the docked decameric complex of WT α-Synuclein (Fig. 4c).
The radius of gyration (Rg) for x (Rgx), y (Rgy) and z-axis (Rgz) were observed to be varying between 1.67 nm and 2.14 nm, whereas the total radius of gyration (Rgt) was found to be constantly stabilizing around 2.3 nm, thus hinting further towards the stability of studied structure of decameric docked complex of WT α-Synuclein (Fig. 4d).
Conclusion. The present in-silico work was undertaken to identify the main therapeutic target protein responsible for PD and the effect of mutations on its pathophysiology. The study revealed α-Synuclein as the main target protein, which is a highly dynamic and disordered protein, existing in multiple polymorphs. Therefore, the common kernel structure of the α-Synuclein was selected to study different oligomerization patterns.
The protein-protein docking study for self-aggregation of five α-Synuclein mutations (A53T, A53E, E46K, G51D and H50Q) revealed that there was a significant change in the binding energies of SNPs in comparison to WT α-Synuclein on oligomerization. 3 SNPs – A53T, E46K and H50Q demonstrated lower binding energies in the decamer stage of oligomers, indicating higher affinities and higher rates of self-aggregation, while 2 SNPs – A53E and G51D displayed higher binding energies in the decameric form, inferring lower affinities and lower rates of aggregation in comparison to WT α-synuclein. G51D and E46K mutated oligomeric structures of α-Synuclein showed twisted morphologies, which may cause a decrease or increase in the rates of aggregation depending upon the structural changes in the active site of α-Synuclein protein. The decameric form of WT α-Synuclein was observed to be stable throughout the molecular dynamics simulation study of 100ns thus reflecting towards its stability. The present study provides a good platform for further investigation to establish α-Synuclein decameric form as a major target protein for PD therapeutic design and development.
Financial support
No financial support has been provided for this work.
Благодарности
the authors wish to thank UGC New Delhi for providing JRF to AG and the High-performance computing (HPC) facility ‘Param Smriti’, NABI, Mohali for the super-computational facility.














Список литературы
Список использованной литературы появится позже.