
Common CNVs modulate phenotypes in neurodevelopmental disorders (autism and intellectual disability): focus on SHANK3 and CHAMP1
Aннотация
Background: Common copy number variations (CNVs) are rarely considered as causative in genomic research dedicated to uncovering mechanisms of neurodevelopmental diseases. However, analyzing complex systems of interactions between individual genomic variations indicates that at least minimal phenotypic effects of common CNVs should exist. Here, we have tested this idea focusing on CNVs affecting SHANK3 and CHAMP1 genes. The aim of the study:The analysis of common CNVs affecting SHANK3 and CHAMP1 genes in a neurodevelopmental cohort (intellectual disability, autism, epilepsy) to uncover possible effects on the phenotypic outcome. Materials and methods: CNVswere evaluated in aneurodevelopmental cohort of 780 children with intellectual disability, autism, epilepsy and congenital anomalies by molecular karyotyping using a high-resolution SNP array technique. Original bioinformatic methods were used to address the effect of CNVs affecting SHANK3 and CHAMP1. Results: CNVs involving SHANK3 and CHAMP1 were observed in 44 (5.6%) and 6 (0.8%) individuals, respectively. SHANK3 CNVs have been associated with specific language problems in 36 (82%) out of 44 individuals. CHAMP1 CNVs were all associated with chromosomal instability, which had been proposed as a cause of more severe clinical manifestations. Bioinformatic analysis has confirmed these modulating effects of common CNVs on phenotypes in neurodevelopmental diseases. Conclusion: Our data allowed to propose a pathogenetic model for brain disfunction in ID and ASD, which is based on an idea that common CNVs add/exacerbate phenotypic traits produced by the main genetic defect (chromosomal aberration or gene mutation). Thus, it appears that mechanisms of neurodevelopmental diseases are more complex than previously recognized even in cases associated with a well-described genomic pathology
Ключевые слова: copy number variations, autism, intellectual disability, neurodevelopmental disorders, phenotype, SHANK3, CHAMP1
К сожалению, текст статьи доступен только на Английском
Introduction. Copy number variations (CNVs) are a major contributor to the pathogenesis of brain diseases. Moreover, it appears that psychiatric (neurodevelopmental) disorders are stronger associated with CNVs than other morbid conditions. This is a special case of autism or autism spectrum disorders (ASD) and intellectual disability (ID) [1, 2]. However, direct associations between ASD/ID and CNVs are generally limited to microdeletion/microduplication syndromes (autism and ID are considered hallmark symptoms) or rare conditions reported as individual cases or occasional findings in cohort studies [1-4]. As a result, the phenotypic effect of CNVs uncovered during genomic analysis of individuals suffering from brain diseases remains a matter of conjecture. Earlier, to solve the causation problem occurring during analyses of CNVs, a variome (CNVariome) concept was applied. Accordingly, it was proposed that both common and rare CNVs are able to produce a cumulative effect on the phenotype [3]. Consequently, it was hypothesized that common CNVs rather possess modulating phenotypic effects than cause the disease per se [3, 4]. To uncover these effects on phenotypes in neurodevelopmental (psychiatric) disorders, specific bioinformatic or systems biology methods are to be applied; fortunately, these are available [4, 5]. On the other hand, there is still a conundrum of interpreting genomic data in the pathogenetic context in neurodevelopmental diseases (e.g., ASD) [6]. The large number of methods for analyzing functional consequences of CNVs hinders the comparative analysis between individual data and case-control studies. However, there are ways to come to a conclusion about pathogenetic value of a variant regardless of occurrence among healthy or affected individuals [7]. For instance, modulating functional consequence of a variant using approaches of system genomics appears to be an issue in understanding the effect of CNVs on phenotype [3, 5, 7]. In total, it seems that a need for a re-evaluation of common CNVs in neurodevelopmental diseases exists.
As previously noted, there are still debates concerning genetic causation in neurodevelopmental disorders (e.g., autism) [6]. However, CNVs are known to be involved in the development of psychiatric symptoms and cognitive ability, especially, those observed in ASD and other childhood psychiatric disorders [8]. CNVs cause changes of protein structures through loss or copy increase of coding sequences. These changes underlie functional consequences of CNVs [9]. Consequently, in silico (bioinformatic) methods based on systems biology analysis and simulating functional consequences of CNVs may be used for uncovering variants possessing phenotypic outcomes [5, 10]. Currently, the impact of CNVs on brain functioning resulting in specific neurobehavioral phenotypes and psychiatric diseases is recognized [11]. Finally, data on CNVs correlates with findings of neuroimaging studies in children with ASD and ID [12, 13]. One may assume that analysis of common CNVs affecting genes, which are defective (gene mutations/CNVs/chromosome imbalances) in syndromic ASD and/or ID, is likely to result in uncovering a less specific, albeit appreciable effect on neurodevelopmental phenotypes.
Previously, we reported a case of partial SHANK3 duplication associated with specific phenotype (neurophysiological biomarker), which appears to contribute to the pathogenesis of ASD with/without ID and other neurobehavioral disorders [12]. SHANK3 encodes one of SHANK postsynaptic scaffolding proteins, which are present at glutamatergic synapses in the brain and are associated with genetic (rare) forms of ASD [14]. Furthermore, heterozygous deletion of SHANK3 gene due to 22q13.3 microdeletion is an important element of pathogenesis of Phelan-McDermid syndrome, which has long been seen as ‘a rare genetic form of autism’ [15]. The involvement of SHANK3 gene in a microdeletion syndrome along with other co-localized genes supports the hypothesis concerning phenotype modulating by SHANK3 copy number change in individuals with other genetic defects and neurodevelopmental diseases. A similar story has emerged in rare neurodevelopmental disorder caused by mutations/CNVs of CHAMP1 (CHAMP1-related disorder) with the only exception of a wide spectrum of neurodevelopmental phenotypes and congenital anomalies (CA) [16]. We hypothesize that common CNVs of these genes, which are generally considered as benign, could possess a modulating effect on neurodevelopmental phenotypes. Here, we have evaluated CNVs detected by molecular karyotyping the high-resolution SNP array technique in the neurodevelopmental cohort reported previously [17] with special attention to common copy number changes of SHANK and CHAMP1 and their phenotypic effects.
The aim of the study. The aim of this study is to describe common CNVs affecting SHANK3 and CHAMP1 genes in a neurodevelopmental cohort of children with ASD, ID, epilepsy and CA and to uncover probable effects on the phenotypic outcome.
Materials and methods. Еру neurodevelopmental cohort of 780 children with ASD, ID, epilepsy and/or neurobehavioural disorder and CA was analysed using molecular karyotyping and a SNP array platform (CytoScan HD Arrays by Affymetrix, Santa Clara, CA). This platform has nearly 2.7 billion markers and nearly 750,000 SNPs for analysing CNVs and heterozygosity losses [17]. CNVs affecting SHANK3 and CHAMP1 were evaluated by original in silico (bioinformatic) methods based on system biology analysis and simulating functional consequences of a genomic variant at transcriptome, proteome/interactome and metabolome levels as systematically described earlier [3, 10, 17, 18]. Interactome visualization and analysis of SHANK3 and CHAMP1 proteins were performed using STRING DB, https://string-db.org/ [19].
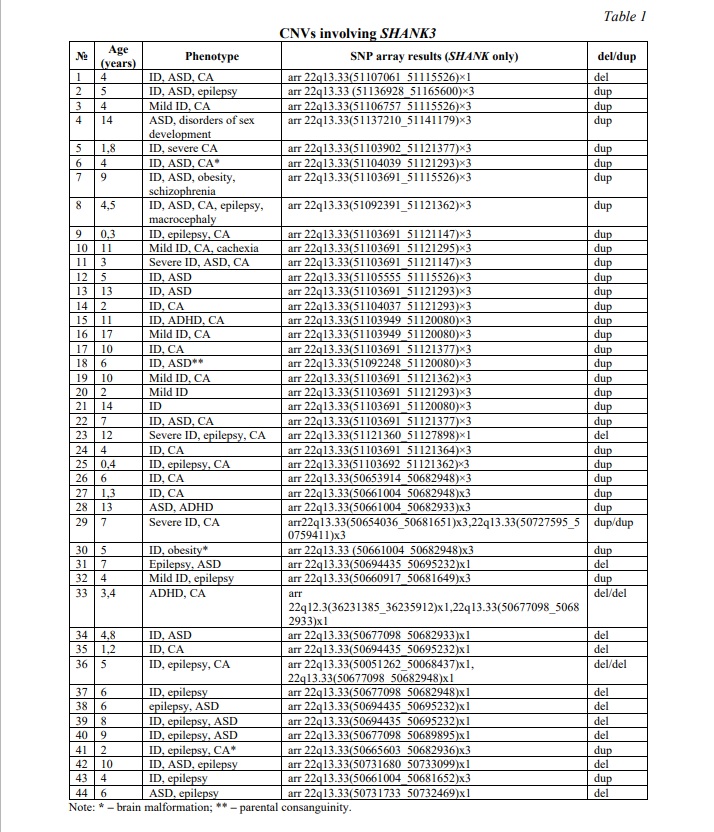
Results and discussion. CNVs affecting the SHANK3 gene were identified in 44 (5.6%) out of 780 children from the neurodevelopmental cohort. Deletions were observed in 13 individuals (1.7%), whereas duplications were found in 31 individuals (3.9%). The higher occurrence of duplications probably indicates that they have a less adverse effect on SHAK3 protein function than deletions do. Table 1 summarizes data on common CNVs involving SHANK3 and phenotypes of the affected individuals. Figure 1A demonstrates examples of CNVs affecting the SHANK3 gene.

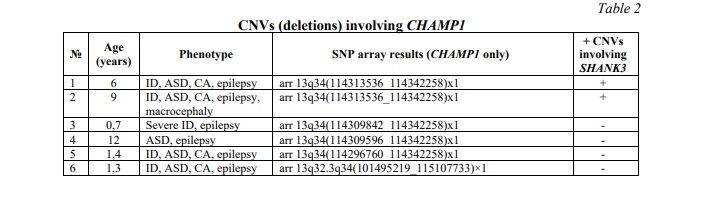
CNVs affecting the CHAMP1 gene were identified in 6 (0.8%) out of 780 children from the neurodevelopmental cohort. All the CNVs were deletions. Two cases demonstrated co-occurrence of SHANK3 duplication and CHAMP1 deletion with severe manifestations of neurodevelopmental disorders and CA. Table 2 summarizes data on common deletions involving CHAMP1 and phenotypes of the affected individuals. Figure 1B demonstrates an example of CHAMP1 deletion.
Children from the neurodevelopmental cohort, generally, shared ASD and ID. CA and epilepsy were common in cohort members with CNVs involving SHANK3 and CHAMP1. Occasionally, children exhibited attention deficit hyperactivity disorder (ADHD), obesity or brain malformations. Certainly, the underlying cause of phenotypes in these children was the result of previously detected defects in chromosomal structure and genes (unshown data; partially presented in [17]). To focus on the possible modulating effect of common CNVs of SHANK3 and CHAMP1, the remaining load of CNVs (chromosomal imbalances and intragenic CNVs) was disregarded, and shared phenotypic traits (e.g. specific neurobehavioural changes, as found in a previous case [12]) were addressed. Earlier, SHANK3 CNVs have been associated with deficient temporal resolution of the auditory system. This led to specific language problems and had been suggested to represent a biomarker of SHANK3 copy number changes. Here, 36 (82%) out of 44 individuals with SHANK3 CNVs exhibited this phenotypic trait. The remainder (patients 5, 6, 10, 11, 23, 29, 30, 41 from Table 1) was associated with extremely severe clinical manifestations of ID and CA with brain malformations, which are likely to mask the modulating effect of SHANK3 CNVs. CHAMP1 CNVs were all associated with chromosomal instability in affected children (unshown data), which had been proposed as a cause of more severe manifestation of ASD, ID, epilepsy and CA. To further demonstrate the modulating effect, a bioinformatic analysis was performed. Figures 2 and 3 demonstrate interactomes of SHANK3 and CHAMP1, respectively.
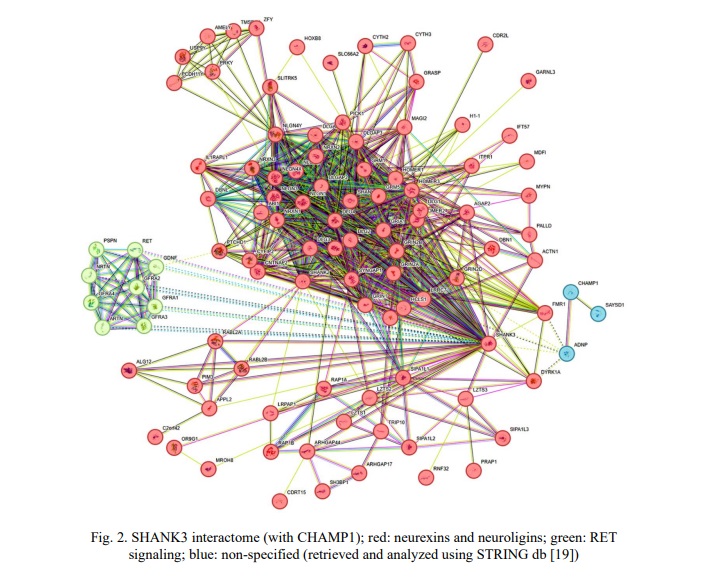
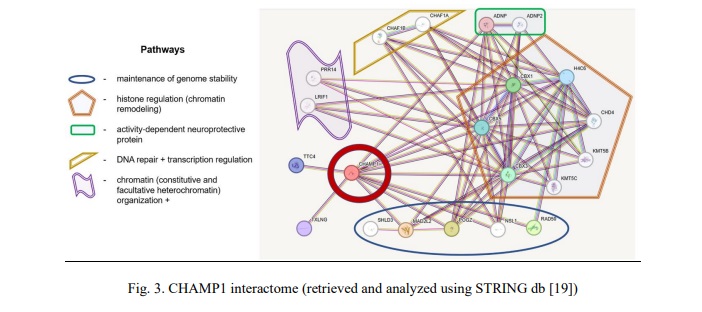
Interactome analysis has not highlighted a valuable network between SHANK3 and CHAMP1. Accordingly, common CNVs of the SHANK3 and CHAMP1 have been considered as phenotype modulators through unshared pathways. SHANK3 is a key element of pathways regulating synaptogenesis (neurexins and neuroligins) and RET signaling. CHAMP1 is a connecting link between pathways involved in chromatin remodeling (histone regulation and heterochromatin formation), maintenance of genome stability (including DNA repair) + transcriptional regulation, and activity‐dependent neuroprotective proteins.
All the CNVs involving SHANK3 and CHAMP1 affect exons leading, therefore, to a change of protein structure, which should influence the phenotype [9] (especially, neurobehavioral phenotype [11]). Furthermore, CNVs of disease-associated genes are linked to specific psychiatric symptoms and cognitive ability (these are phenotypic traits, but not disorders!) [8] and neurophysiological endophenotypes [13]. Additionally, genomic studies of synaptic dysfunction in ASD have revealed that specific phenotypic traits result from mutations in 'postsynaptic' genes, including SHANK3 [20]. In the present study, specific language problems (for more details, see [12]) have been associated with CNVs involving SHANK3 in 82% of cases. It should be noted that the remaining cases exhibited extremely severe ID and CA, making it impossible to evaluate speech impairment. Children without CNVs of the SHANK3 gene have not demonstrated this specific language/speech problems. Similar changes have been reported during genotype-phenotype correlations in individuals with SHANK3 de novo intragenic mutations [21]. Furthermore, SHANK3 deficiency is found to result in detectable pathological processes in the central and peripheral nervous system [22]. As shown by genetic model studies [23, 24] and SHANK protein expression analysis [25], despite a wide spectrum of SHANK3 ontologies and abundant distribution (expression), alterations to the sequence and copy number of this gene is almost always associated with specific phenotypic and endophenotypic changes. These observations are relevant to data retrieved from the interactome and further bioinformatic analysis. Therefore, the modulating effect of CNVs affecting the SHANK3 manifesting as specific language/speech problems is highly probable. No correlations between phenotypic outcomes and type/localization of CNVs have been determined.
Rare microdeletions at 13q33q34 (CHAMP1 gene localization) and mutations in CHAMP1 gene have long been associated with a disorder exhibiting a wide spectrum of neurodevelopmental/neurobehavioral abnormalities (autism, ADHD and ID) [26, 27, 28]. Deficiency of this gene causes neurodevelopmental impairment manifesting as more-or-less specific neurobehavioral phenotypes [28]. Functionally, CHAMP1 represents an important cellular component, alterations to which seem to produce abnormal chromatin remodeling (transcription regulation and homologous recombination included) and neuroprotection [27, 29]. Additionally, functional genomic studies of CHAMP1 complex have demonstrated that it is strongly required for maintaining genomic stability in euchromatic and, more specifically, heterochromatic regions [30]. The failure of the latter leads to genome and chromosome instability, which have been systematically associated with a wide spectrum of brain diseases, including ASD and epilepsy, as a key element of the pathogenetic cascade [31, 32, 33]. Since both bioinformatic analysis (Fig. 3) and literature data provide evidence of alterations to CHAMP1-realted pathways resulting in failures to genome/chromosome stability maintenance, we have concluded that chromosomal instability mediated by CHAMP1 CNVs (deletions) is the driving force for modulating the phenotype or leading to more severe forms of ID, ASD and CA.
Despite the knowledge about the role of CNVs in interindividual diversity and pathophysiology of brain diseases, there is still a gap in our understanding of mechanisms, by which copy number changes of genes produce neurodevelopmental phenotypes. In general, it is suggested that CNVs produce a phenotype in one of two ways: either directly (i.e. by altering the structure or copy number of protein-coding sequences of a disease-causing gene) [9, 11], or via the cumulative effect of CNVs (i.e. the CNVariome, or a complex system of interactions between CNVs and genes/proteins changed by CNVs) [3]. At the neurophysiological level, CNVs are able to cause endophenotypic traits, which are specific to a given disease-causing gene [12, 34]. Moreover, specific phenotypic traits of neurodevelopmental disorders (psychiatric symptoms, cognitive disability, etc.) are associated with CNVs involving disease-causing genes [8]. Regardless of the focus on rare CNVs as a cause of neurodevelopmental and neurobehavioral diseases [2, 11], almost all these observations are applicable to common CNVs with the sole exception of diminished phenotypic effect of common CNVs, probably due to natural/evolutionary selection. Thus, common CNVs are to be considered as valuable contributors to phenotypes of neurodevelopmental diseases (ASD and ID).
In total, the involvement of common CNVs appears to be described by the following model. The main genetic defect (e.g., gene mutation or chromosomal aberration) produces the essential clinical picture of the disease (a combination of hallmark symptoms), whereas common CNVs add or exacerbate phenotypic traits (especially, neurobehavioral phenotypic traits). It is important to state that common CNVs are not the cause of the disease per se. Undoubtedly, this idea requires additional experiments to be ultimately supported.
Conclusion. This study provides evidence that CNVs are important for neurodevelopmental diseases. The purpose of our communication was not to provide a new genetic cause of brain diseases. Indeed, we have proposed a model for brain disfunction in ID and ASD, which is based on an idea that common CNVs add/exacerbate phenotypic traits resulting from the main genetic defect (chromosomal aberration or gene mutation). Here, a support of this idea appears to be presented. Certainly, additional case-control studies of CNVs involving SHANK3, CHAMP1 and other genes are required to identify the intrinsic role of common CNVs in ID, ASD, ADHD and epilepsy. Finally, we conclude that the mechanisms underlying neurodevelopmental diseases are far more complex than was previously recognised, even in cases associated with a well-described genomic pathology.
Financial support
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (the Federal Scientific and Technical Programme for Genetic Technologies Development for 2019-2030, Agreement № 075-15-2025-474.














Список литературы
Список использованной литературы появится позже.