
Inhibitors of intracellular signaling affect the production of soluble endoglin by JEG-3 trophoblast cells upon interaction with NK cells
Aннотация
Background: The cytotoxic activity of natural killer (NK) cells is essential for the proper invasion of trophoblasts into the uterine mucosa. Endoglin (Eng) plays an important role in the interaction between NK cells and trophoblasts by assembling the TGFβ receptor complex. The formation of soluble forms of (s)MICA and (s)MICB can help evade NK cell cytotoxicity. The aim of the study: to assess the interaction between NK-92 line NK cells and JEG-3 trophoblast cells via soluble forms of (s)Eng, sMICA and sMICB, when using a TGFβ-dependent signaling pathway inhibitor and a cyclin-dependent kinase (CDK) 7 inhibitor. Materials and methods: We used NK-92 line cells and JEG-3 line trophoblast cells, the TGFβRI inhibitor LY3200882, and the CDK7 inhibitor THZ1. We incubated cells with LY3200882 or THZ1, and then cultured cells with inducers TNFα, IL-10, IFNγ, TGFβ or trichostatin A (TSA). After 24 hours, we assessed the concentrations of sEng, sMICA, sMICB in the medium. Results: We showed that LY3200882 decreased sEng production in JEG-3 line cells. Cytokines IL-10, IFNγ, and TGFβ abolished the effects of LY3200882 on sEng production by JEG-3 line cells. LY3200882 also induced sMICA formation by NK-92 line cells. THZ1 reduced sEng production by JEG-3 line cells, both in the absence of cytokines and following TNFα, IL-10, IFNγ, and TGFβ stimulation. When co-cultured JEG-3 line cells with NK-92 line cells, THZ1 also lowered sEng levels. In the presence of TSA, JEG-3 cells increased sMICB production. However, after exposure of JEG-3 line cells to THZ1 and TSA, we observed a decrease in sMICB levels. Conclusion: Both the TGFβ-dependent signaling inhibitor and the CDK7 inhibitor influence the interaction of NK-92 line cells with JEG-3 line trophoblast cells, affecting sEng production by JEG-3 line cells. The regulation of sEng formation by trophoblast cells presents new therapeutic avenues for managing reproductive pathologies
К сожалению, текст статьи доступен только на Английском
Introduction. Natural killer cells (NK cells) play an important role in regulating placental formation and, consequently, the normal course of pregnancy. Changes in their activation status are linked with reproductive disorders, such as recurrent miscarriage (RM) in the first trimester of pregnancy and preeclampsia (PE) in the third trimester [1]. NK cells are also key players in the anti-tumor immune response. However, during tumor progression, influenced by the cellular microenvironment, NK cells undergo phenotypic and functional changes, becoming similar to decidual NK cells in the uterus [2]. Studying NK cells is particularly relevant for developing targeted anti-tumor therapies [1, 2]. Due to the similarities between decidual NK cells and tumor-infiltrating NK cells, therapeutic strategies used for treatment of oncological diseases could potentially be adapted for threating reproductive pathologies [1].
The cytokine TGFβ is an important regulatory factor that influences NK cell functional activity in the uterus. Notably, tumor cells actively produce TGFβ [2]. For example, activation of the TGFβ-dependent signaling pathway was observed in cervical cancer [3]. TGFβ is secreted in an inactive form, and its release from the inactivating peptide complex is necessary for signal transmission. The active form of TGFβ binds to a TGFβ receptor (TβR) II dimer, then to a TβRI dimer, forming a heterotetrameric complex [4]. There are three isoforms of the cytokine TGFβ: TGFβ1, TGFβ2, TGFβ3, all of which are present at the uteroplacental interface. TGFβ1 and TGFβ3 can directly bind to TβRII, with subsequent attachment of the co-receptor endoglin (Eng). Binding of TGFβ2, however, requires Eng for binding to TβRII [5, 6].
Eng is involved in activating different signaling pathways initiated by the assembly of the heterotetrameric TGFβ receptor complex. For example, phosphorylation of its membrane form (L-Eng) results in activation of the Smad1/5/8 signaling pathway. The short form of Eng (S-Eng), containing a short cytoplasmic tail, binds to TβRI and triggers Smad2/3-dependent signaling [7]. A soluble form of Eng (sEng), generated through metalloproteinase 14 proteolytic activity, can bind TGFβ and compete for its bioavailability [6]. Normally, Eng is expressed by trophoblast cells, however, high concentrations of sEng are observed in pregnant women with PE [6].
The interaction between NK cells with trophoblast cells involves the participation of Eng [8]. Blocking TGFβ-dependent signaling may potentially regulate this interaction. For example, in RM, the expression of the protein EHD1 – a protein that regulates endocytosis of plasma membrane proteins – increased in trophoblast cells [9]. Experimentally demonstrated that EHD1 induces apoptosis of HTR8 trophoblast cells and reduces trophoblast invasion in mice, leading to impaired spiral artery remodeling [9]. Inhibition of TGFβ-dependent signaling by LY3200882 suppressed the effects of EHD1, in particular reducing pSMAD2 levels [9]. X. Wu et al. concluded that the EHD1-TGFbR1-SMAD2/3 pathway could be a therapeutic target for overcoming RM [9]. However, the impact of the TGFβRI inhibitor LY3200882 on sEng production within the NK cell-trophoblast system remains poorly studied.
In the context of developing antitumor drugs, inhibitors of cyclin-dependent kinase (CDK) 7 – a protein that regulates gene transcription and the activity of other cyclin-dependent kinases within the RNA polymerase complex – are under investigation. CDK7 is known to stimulate the phosphorylation of CDK1 and CDK2, thereby participating in cell cycle regulation [10]. The disruption of this regulation can contribute to malignancy. The selective CDK7 inhibitor THZ1 is used to block tumor cell proliferation [11]. TGFβ induces tumor cell resistance to antitumor therapy with CDK7 inhibitors [11], although THZ1 also suppresses TGFβ-dependent signaling [12]. It is important to note that TGFβ also affects NK cells during cancer progression. For example, activation of the TGFβ signaling pathway via the TGFβ3-TβRI/II interaction was observed in fibroblasts, T lymphocytes, and NK cells in cervical cancer [3]. However, the effects of THZ1 on NK cells, including in the microenvironment of TGFβ-secreting cells, has not been previously studied.
NK cells can recognize target cells, including tumor cells of various origins, by binding the NK cell activation receptor NKG2D to proteins of the major histocompatibility complex, MICA and MICB [13, 14]. However, target cells can evade NK cell cytotoxicity by shedding MICA and MICB [13, 14]. The uterine decidua contains mesenchymal stromal cells expressing MICA and MICB, which can be targeted and lysed by activated NK cells [15]. Soluble forms of (s)MICA, (s)MICB found in cord blood plasma can alter the cytotoxic activity of NK cells in vitro [16]. In anti-tumor therapy, strategies aimed at reducing the levels of sMICA and sMICB and enhancing NK cell cytotoxicity are used [14]. Given the possibility of contact between NK cells and trophoblast cells in the uterus, it is important to determine their expression of sMICA and sMICB, as well as the impact of intracellular signaling inhibitors on these cells.
The aimof the study was to evaluate the interaction of NK-92 line NK cells with JEG-3 line trophoblast cells via sEng, sMICA and sMICB in the presence of an inhibitor of the TGFβ-dependent signaling pathway and an inhibitor of the cyclin-dependent kinase CDK7.
Materials and Methods. Cell lines. We used the NK-92 cell line (ATCC, USA), which exhibits the primary phenotypic, morphological and functional characteristics of activated natural killers [17]. We cultured the NK-92 line cells in a complete growth medium based on the minimal α-modified Eagle's medium (α-MEM) (Biolot, Russia) supplemented with IL-2 (500 U/ml) (Roncoleukin, Biotech, Russia). We cultured the cells in a humidified atmosphere at 37 ̊°C with 5% CO2 and subcultured them every two days.
We used the JEG-3 cell line (ATCC, USA) for trophoblast cells, as this mirrors the main properties of extravillous trophoblast cells during the first trimester of pregnancy [18, 19]. We cultured the cells in a complete growth medium based on the basic modified Eagle's medium (DMEM) (Biolot, Russia), in a humidified atmosphere at 37 °C with 5% CO2 concentration. The cells were subcultured every 3-4 days.
Inductors and inhibitors. To inhibit TβRI signaling, we used the inhibitor LY3200882 at a concentration of 10 nmol/ml (Selleck Chemicals, USA). For selective transcription inhibition, the CDK7 kinase inhibitor THZ1 was used at a concentration of 1 nmol/ml (Selleck Chemicals, USA). We determined the concentrations of these inhibitors based on the manufacturer's recommendations and literature data [20, 21]. We chose the exposure duration for LY3200882 following the guidelines in the literature [22, 23].
The cytokines used as inducers included IL-10, IFNγ, TGFβ, and TNFα, at the following working concentrations: IL-10 (10 ng/ml), IFNγ (1000 IU/ml), TGFβ (5 ng/ml), and TNFα (50 U/ml) (all from R&D Systems, USA). Additionally, we utilized trichostatin A (TSA), a specific histone deacetylase inhibitor, at a working concentration of 1 μg/ml (Merck, USA). We chose the cytokine concentrations in accordance with the manufacturers' recommendations. All stock solutions were prepared based on Hanks' solution (Biolot, Russia).
Cultivation of JEG-3 line trophoblast cells with NK-92 line cells in the presence and absence of cytokines and inhibitors. We seeded JEG-3 line trophoblast cells into a 24-well adhesion plate at a concentration of 200,000 cells per ml. After 24 hours in culture, they formed a monolayer. The following day, we centrifuged the plate at 100×g for 3 minutes. After removing the medium, we added 500 μl of NK-92 line cell suspension (600,000 cells per ml) to the wells with the trophoblast cells (hereinafter referred to as co-culture). Additionally, we transferred some of the NK-92 cells to the wells that did not contain JEG-3 line trophoblast cells (hereinafter referred to as NK-92 cell monoculture). We also cultured some of the JEG-3 line cells without the addition of NK-92 line cells (hereinafter referred to as JEG-3 cell monoculture).
Subsequently, we added the LY3200882 inhibitor to some wells with NK-92 cells, JEG-3 cells, or the co-culture and incubated them for 24 hours. Four hours prior to the end of incubation, we added the THZ1 inhibitor to other wells containing NK-92 cells, JEG-3 cells, or the co-culture, thus exposing the cells to this inhibitor for 4 hours.
After incubation, we transferred the culture medium containing the cells into Eppendorf tubes and centrifuged them at 200×g for 5 minutes. The medium was then removed, and we added 500 μl of Hanks' solution to the Eppendorf tubes, thus washing the NK cells from the inhibitors. Similarly, we removed inhibitors from the trophoblast cells by adding 500 μl of Hanks' solution to the wells and centrifuging the plates at 200×g for 5 minutes. After disposing of the Hanks' solution, 500 μl of complete growth medium for NK cells was added to the Eppendorf tubes with the NK cells. We returned the contents of the Eppendorf tubes to the corresponding wells in the plate. We then incubated the cells for 24 hours, adding inducers (IL-10, IFNγ, TGFβ and TNFα) in combination with each inhibitor, as well as in the absence of inhibitors. Some cells were left without inhibitors or inducers as a control. Four independent experiments were conducted (n = 4), with two technical replicates in each.
After incubation, we centrifuged the plates with cells at 100×g for 3 minutes and collected the conditioned medium (CM), which we stored at -80 °C for no more than 2 months. The experimental design is illustrated in Fig. 1.
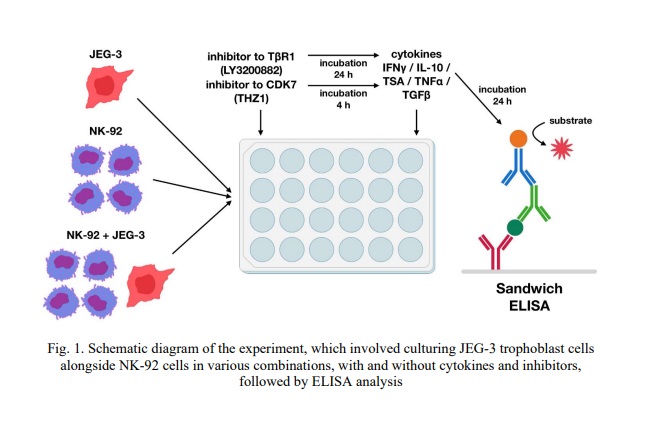
Evaluation of sMICA, sMICB, sEng secretion by JEG-3 line trophoblast cells and NK-92 line cells. For the two-center enzyme-linked immunosorbent assay (ELISA), we used a kit developed by the A. Granov Russian Research Center for Radiology and Surgical Technologies, Hybridoma Technology Laboratory [24]. The kit included monoclonal antibodies (MAB) against MICA (clones IA4+4AII/, IA4+2GI, 10 ng/ml), MICB (clones 4DII+4E3+2A3/1B8+2A8, 2.5 ng/ml), Eng (clone 4E4/4C9, 20 ng/ml); MAB for sorption; calibrator solution (R&D Systems, USA); Tris-Tween solution; TMB solution; reaction stop solution; phosphate saline solution (PSS). We also used 96-well Nunc MaxiSorp plates (Thermo Fisher Scientific, USA).
We assessed the secretion of sMICA, sMICB and sEng proteins in accordance with the manufacturer's instructions. We measured the optical density at a wavelength of 450 nm using an ELx808 spectrophotometer (Biotek Instruments, USA).
Statistical processing. We performed the statistical analyses using the GraphPad Prism 8 programme (GraphPad Software, USA). For paired comparisons, we used the non-parametric Mann–Whitney rank test. For multiple comparisons, we used the Kruskal-Wallis test with Dunn's post hoc test. We defined differences as statistically significant at p<0.05. We analysed samples preliminarily for outliers.
The study adhered to the Code of Medical Ethics of the World Medical Association (Declaration of Helsinki). The Local Ethics Committee of D.O. Ott Research Institute of Obstetrics, Gynecology, and Reproductology approved the research protocol (Protocol No. 135 dated 30th May 2024).
Results and discussion
sEng content in the CM of NK-92 and JEG-3 cell lines after their incubation with the TGFβ-dependent signaling pathway inhibitor LY3200882 and the cyclin-dependent kinase inhibitor CDK7 THZ1. To present the results more clearly, we prepared the graphs comparing the sEng concentration in the CM of the JEG-3 cell line monoculture after exposure to LY3200882 (Fig. 2A) and THZ1 (Fig. 2B). Figure 3 illustrates the comparison of sEng levels in the monocultures of NK-92 and JEG-3 cells, as well as in their co-culture. Additionally, the sEng content in the co-culture CM is presented in the presence of LY3200882 (Fig. 4A) and THZ1 (Fig. 4B).
Our findings indicate that sEng is present in the CM of JEG-3 cells. After incubation with the inhibitor LY3200882, the sEng concentration in the CM of the JEG-3 cell monoculture reduced compared to that in the CM of intact JEG-3 cells (Fig. 2A). We demonstrated that culturing JEG-3 cells with TSA resulted in a decrease in sEng concentration in the CM, whereas cytokines had no effect on sEng levels (Fig. 2). Furthermore, the sEng concentration in the CM of JEG-3 cell monocultures that were first treated with LY3200882, followed by TSA or the cytokine TNFα, was reduced compared to monocultures that were solely treated with TSA or TNFα (Fig. 2A).
We demonstrated that the concentration of sEng in the CM of JEG-3 cells treated with the THZ1 inhibitor was lower than in intact JEG-3 cells (Fig. 2B). THZ1 was found to maintain its inhibitory effect on sEng levels in the CM of the JEG-3 cell monoculture even when cytokines such as TNFα, IL-10, IFNγ and TGFβ were present in the media (Fig. 2B).
In the CM of the NK-92 cell monoculture, sEng was absent after incubating the cells without inducers, after pretreating them with the inhibitors LY3200882 and THZ1, and during cultivation with all the inducers used in the study (see Fig. 3).
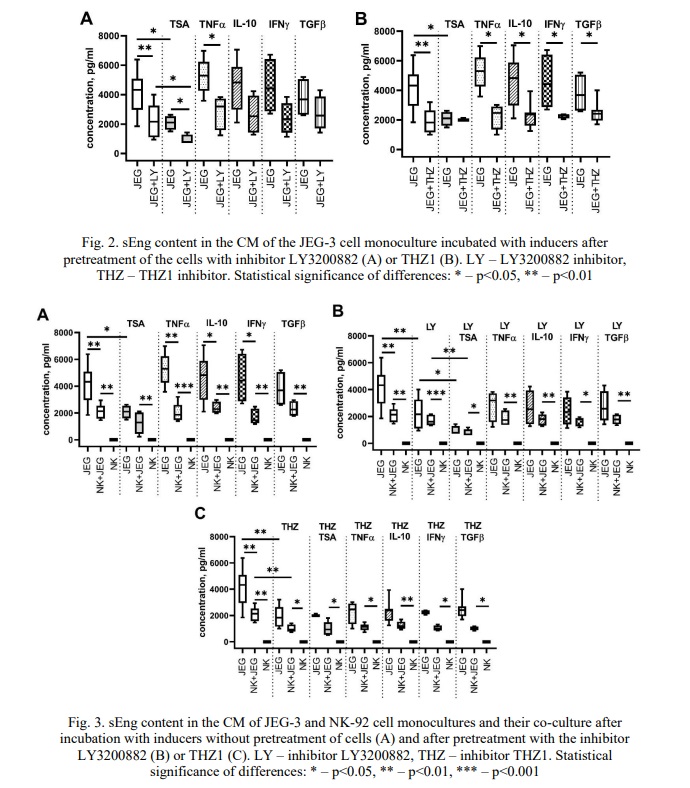
We detected sEng in the CM from the co-culture, in contrast to the CM obtained from the NK-92 cell monoculture (Fig. 3A-B). This increase in sEng levels was observed both in the CM from cells cultured without inhibitors and in the CM from cells treated with the inhibitors LY3200882 (Fig. 3B) and THZ1 (Fig. 3C). Notably, cytokines and TSA did not alter the sEng concentration in the co-culture CM (Fig. 3A).
We observed that the concentration of sEng in the CM from the co-culture was lower than in the CM from the JEG-3 cell monoculture incubated without inducers (see Fig. 3). A similar trend was observed when cytokines TNFα, IL-10 and IFNγ were present, but not TGFβ: the sEng concentrations in the co-culture CM were lower than in the JEG-3 cell monoculture (Fig. 3B). Furthermore, the sEng concentration in the CM of the co-culture did not differ from that of the JEG-3 cell monoculture in the presence of TSA (Fig. 3A).
Pre-incubation with the inhibitor LY3200882 had no effect on the concentration of sEng in the CM of the JEG-3 cell monoculture or the co-culture (Fig. 3B), regardless of whether the cells were subsequently incubated with cytokines. TSA decreased the sEng concentration in co-culture CM pretreated with LY3200882 (Fig. 3B, 4A).
Similarly, pre-incubation with THZ1 reduced the sEng concentration in the CM of the JEG-3 cell monoculture, resulting in no difference compared to the co-culture (Fig. 3B). Comparisons of sEng concentrations in the co-culture indicated that THZ1 resulted in decreased sEng levels compared to the co-culture without the inhibitor (Fig. 3B, 4B).
We found that the inhibitor LY3200882 had no effect on sEng levels in the co-culture CM, either during incubation without cytokines or after culturing in a medium containing cytokine (Fig. 4A).
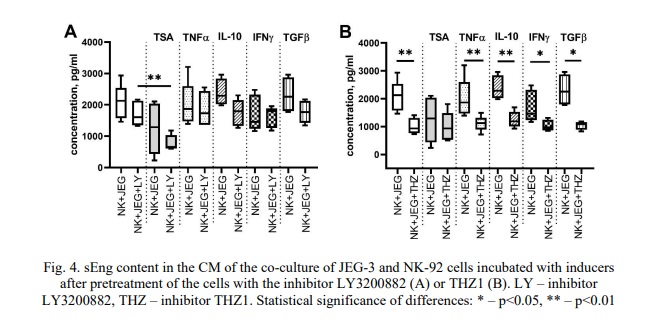
The inhibitor THZ1 reduced the sEng concentration in the co-culture CM compared to the co-culture CM incubated without THZ1 (Fig. 4B). This effect maintained in the presence of the cytokines TNFα, TGFβ, IL-10, IFNγ, but not TSA (Fig. 4B).
sMICA content in the CM of NK-92 and JEG-3 cell lines after their incubation with the TGFβ-dependent signaling pathway inhibitor LY3200882 and the cyclin-dependent kinase inhibitor CDK7 THZ1. We observed that both JEG-3 and NK-92 cell monocultures did not produce sMICA, regardless of whether they were cultured in the presence or absence of inducers. The addition of the inhibitor LY3200882 or THZ1 did not lead to the detection of sMICA in the CM of trophoblast cells (Fig. 5).
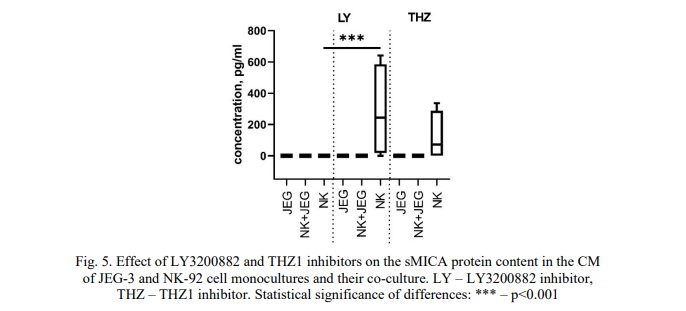
However, the inhibitor LY3200882 resulted in an increased level of sMICA in the NK-92 cell monoculture compared to the CM from intact NK cells (Fig. 5). Furthermore, after treating NK cells with LY3200882 and subsequently adding inducers, we did not detect any sMICA proteins in the CM. Similarly, no sMICA was identified in the CM of the co-culture.
sMICB content in the CM of NK-92 and JEG-3 cell lines after their incubation with the TGFβ-dependent signaling pathway inhibitor LY3200882 and the cyclin-dependent kinase inhibitor CDK7 THZ1. We detected sMICB in the CM of the JEG-3 cell monoculture (Fig. 6). The addition of cytokines TGFβ, IFNγ, TNFα, IL-10 did not significantly alter the concentration of sMICB in the CM of the JEG-3 cells. However, following treatment with TSA, we observed an increase in the sMICB protein levels in the CM of the JEG-3 cells compared to those cultured without the inducer (Fig. 6).
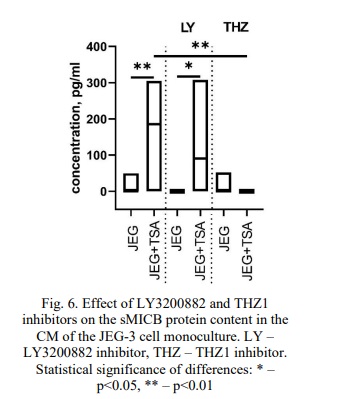
The inhibitors LY3200882 and THZ1 did not affect the sMICB concentration in the CM of the JEG-3 cell monoculture. Notably, we detected a higher level of sMICB in the CM after pretreating JEG-3 cells with LY3200882 and then exposing them to TSA than when the cells were incubated with LY3200882 alone (Fig. 6). While the inhibitor THZ1 did not change the sMICB concentration in the CM of JEG-3 cells without inducers, we found that the sMICB content was lower after treatment with both THZ1 and TSA compared to treatment with TSA alone (Fig. 6).
Additionally, we determined that NK-92 cells do not produce sMICB. No sMICB proteins we detected in the CM from the co-culture of NK-92 and JEG-3 cells. We did not reveal any effect of LY3200882 and THZ1 inhibitors, as well as the inducer TSA and cytokines TGFβ, IFNγ, TNFα, IL-10 on the sMICB content in either the CM of the NK-92 cell monoculture or the CM of the co-culture.
Figure 7 summarizes the main findings of our study.

Overall, we discovered that JEG-3 trophoblast cells produce sEng, and that treatment with TSA leads to a decrease in the concentration of sEng in the CM. Previous studies have shown that JEG-3 cells express Eng on their plasma membrane and generate its soluble form [8]. The production of sEng by trophoblast cells is believed to be a mechanism for evading the cytotoxic activity of NK cells [8]. According to the literature, epigenetic modifications of DNA, including histone deacetylation, influence the migration and proliferation capabilities of both primary trophoblast cells and JEG-3 cells [25, 26]. Exposure to TSA induces histone hyperacetylation, which relaxes DNA and modulates gene expression [27]. Therefore, the observed reduction in sEng production by JEG-3 cells after TSA treatment can be interpreted as a cellular response to a stress factor. Moreover, it is important to consider that sEng formation may not only result from the activity of MMPs but could also arise from epigenetic modifications occurring directly within the trophoblast cells.
The cytokine TGFβ is synthesized by trophoblast cells, including JEG-3 cells [28]. It regulates the invasion of trophoblast cells [29], and a deficiency of this cytokine may lead to miscarriage. In the context of PE, an increased production of TGFβ in the placenta has been observed [30]. Additionally, elevated levels of sEng have been detected in patients with PE [6]. We demonstrated that inhibiting TGFβ-dependent signaling using LY3200882 reduced sEng concentrations in JEG-3 cells. This suggests that sEng production by trophoblast cells is dependent on TGFβ, which may contribute to the pathogenesis of PE, alongside sEng synthesis by endothelial cells.
Following the treatment of JEG-3 cells with either the inhibitor LY3200882 or TSA, we observed a significant decrease in sEng concentrations. This is likely to be the result of the combined effects of TGFβ deprivation and cellular stress. A similar reduction in sEng levels occurred when JEG-3 cells were co-cultured with NK cells.
Furthermore, adding TNFα to JEG-3 cells after treating them with LY3200882 resulted in a further decrease in sEng concentrations in the CM, likely due to TNFα's pro-inflammatory effects. Conversely, the cytokines IL-10, IFNγ and TGFβ counteracted the inhibitory effect of LY3200882 on sEng production by JEG-3 cells. Notably, IL-10 and IFNγ are present at the uteroplacental interface and have been shown to be secreted by decidual NK cells [31]. Prolonged exposure to IL-10 and IFNγ results in stable HLA-G expression in JEG-3 trophoblast cells [32]. Therefore, inhibiting the TGFβ-dependent signalling pathway may have a reduced impact on sEng production in the presence of these cytokines.
At the same time, it is important to highlight the cancellation of the effect of the inhibitor LY3200882 on the formation of sEng in the presence of TGFβ. Previous studies have suggested that sEng can bind TGFβ, functioning as a carrier protein that facilitates the transfer of sEng between cells, particularly from trophoblasts to NK cells [8]. When exogenous TGFβ is added to JEG-3 cells, an excess amount of this cytokine may bind to sEng, inhibiting the detection of sEng by the antibodies used in the ELISA assay. This binding may lead to a decrease in the detectable sEng in the CM and result in no observable differences in sEng levels in JEG-3 cells following TβRI blockade. Overall, further research is needed to clarify the relationship between sEng production by trophoblast cells and the functional activity of TβRI.
We found that JEG-3 cells reduced sEng production upon exposure to the inhibitor THZ1, an effect noted both in the absence of cytokines and after stimulation with TNFα, IL-10, IFNγ, and TGFβ. Notably, THZ1 also resulted in a decrease in sEng concentration in co-culture conditions. These findings suggest that THZ1 exerts a more pronounced effect on trophoblasts, diminishing their ability to regulate recognition by NK cells.
As established previously, NK-92 cells do not produce sEng [8]. In the current study, we demonstrated that coculturing NK-92 cells with JEG-3 cells led to a reduction in sEng content in the CM compared to the levels observed in the CM of JEG-3 cells alone. This finding supports the hypothesis that sEng is involved in the intercellular interactions between NK cells and trophoblasts. We noted these differences in sEng levels both in the absence of cytokines and in the presence of TNFα, IL-10, and IFNγ. However, in the presence of TGFβ, there were no changes in the sEng content in the JEG-3 cell monoculture and their co-culture with NK cells. The results obtained can be explained by our hypothesis regarding alterations in the configuration of sEng epitopes due to TGFβ binding, which may occur when there is an excess of this cytokine in the CM, resulting in decreased sensitivity of the ELISA assay.
We observed that following preliminary incubation of cells with both the TGFβ-dependent signaling pathway inhibitor and the CDK7 inhibitor, the sEng concentration in the CM of the co-culture did not differ from that observed in the JEG-3 cell monoculture. We attribute these results to the decreased sEng level in the CM from JEG-3 cells, which we detected after treatment with THZ1 and LY3200882.
In this study, we also evaluated the impact of inhibitors THZ1 and LY3200882 on the production of sMICA and sMICB by NK-92 and JEG-3 cell lines. The expression of MICA and MICB has been shown in many types of cancer [33]. Shedding of these ligands from the cell membrane may enable cells to evade recognition by cytotoxic lymphocytes and NK cells [34]. Elevated levels of sMICA and sMICB levels are associated with malignancy [35, 36]. Overall, there is currently limited data on MICA/B production by NK cells.
The NK-92 cells used in our experiments were derived from a patient with non-Hodgkin's lymphoma. Immunohistochemical analysis of lymph nodes from patients with this condition shows the expression of MICA and MICB, along with elevated levels of their soluble forms in the bloodstream [36]. A significant increase in the concentration of sMICA and sMICB has been linked with a poor prognosis of this disease [36]. Furthermore, NK-92 cells are known to express MICA and MICB on their membrane [8], suggesting that these cells may also produce sMICA and sMICB.
Our data indicate that intact NK-92 cells did not produce sMICA and sMICB. However, upon exposure to a TGFβ-dependent signaling inhibitor, the concentration of sMICA in the CM from NK-92 cells increased. We have previously established that NK-92 cells secrete TGFβ [37]. It is likely that the blockade of TβRI resulting from incubation with LY3200882 serves as a stress signal for NK cells, prompting them to shed sMICA in an attempt to evade cytolysis.
We found that JEG-3 cells produce sMICB, and its concentration increases in the presence of TSA. These results indirectly support the hypothesis that TSA induces stress in JEG-3 cells. Interestingly, inhibition of TGFβ-dependent signaling did not alter the secretion of sMICB by JEG-3 cells. When CDK7 was inhibited and JEG-3 cells were exposed to TSA, we observed a decrease in sMICB concentration. This may be attributed to TSA induced release of DNA from histone binding. Yet, THZ1 blocks the activity of the DNA polymerase complex. We assume that these opposing mechanisms contribute to the cancellation of the effect of TSA on JEG-3 cells.
Conclusion. Inhibiting TGFβ-dependent signaling with LY3200882 decreased sEng levels in JEG-3 cells. However, the presence of the cytokines IL-10, IFNγ and TGFβ negated the effect of LY3200882 on sEng production in these cells. Furthermore, LY3200882 induced sMICA formation in NK-92 cells, which suggests that these cells avoid cytolysis when deprived of TGFβ signaling.
Furthermore, the CDK7 inhibitor THZ1 decreased sEng production in JEG-3 cells, both in the absence of cytokines and following stimulation with TNFα, IL-10, IFNγ, and TGFβ. In co-culture of JEG-3 cells with NK-92 cells, THZ1 also led to a decrease in sEng concentration. These findings suggest that THZ1 significantly affects trophoblast cells, resulting in a diminished ability to regulate their recognition by NK cells. In the presence of TSA, JEG-3 cells increased the production of sMICB. However, after treatment with both THZ1 and TSA, we observed a decrease in sMICB concentration, which may be linked to the inhibition of the DNA polymerase complex.
Thus, both the TGFβ-dependent signaling inhibitor and the CDK7 inhibitor influence the interaction of NK-92 cells with JEG-3 trophoblast cells, thereby altering the production of sEng by JEG-3 cells.
Financial support
The work was supported by Grant № 25-24-00028 (2025-2026) from the Russian Science Foundation.














Список литературы
Список использованной литературы появится позже.