
Гено-фенотипические особенности муковисцидоза у российских детей из Чеченской и Карачаево-Черкесской Республик
Aннотация
Актуальность: Муковисцидоз (МВ) – это аутосомно-рецессивное заболевание, которое встречается с частотой от 1:1500 до 1:5000 новорожденных по данным Всемирной Организация Здравоохранения. В Российской Федерации средняя частота болезни составляет 1:10000 новорождённых. Медико-социальная значимость данного заболевания связана с ранней инвалидизацией пациентов, необходимостью их многолетнего лечения и постоянного диспансерного наблюдения, а также гетерогенностью фенотипических проявлений, что в свою очередь требует ранней постановки диагноза с помощью молекулярно-генетических методов. Цель исследования:Изучить клинические и молекулярно-генетические особенности детей с МВ из Чеченской и Карачаево-Черкесской Республик. Материалы и методы:В исследование включено 237 пациентов с подтвержденным диагнозом МВ. Молекулярно-генетическая диагностика осуществлялась методом массового параллельного секвенирования c использованием гибридизационной таргетной панели, включающей весь ген CFTR. Секвенирование проводилось на платформе MiSeq. Все каузальные нуклеотидные варианты были валидированы при помощи секвенирования методом Сэнгера. Результаты:Наиболее частыми аминокислотными вариантами гена CFTR у пациентов с МВ из Чеченской Республики являются: p.Y515* (94 аллеля/78,3%), p.E92K (18 аллелей/15%). Наличие в генотипе вариантов p.Y515* и p.E92K в гетерозиготном состоянии чаще является благоприятным прогностическим фактором отсутствия поражения поджелудочной железы. Основной отличительной клинической особенностью данных пациентов, является дебют заболевания с проявлений синдрома псевдо-Барттера. Мажорным аминокислотным вариантом гена CFTR у пациентов с МВ из Карачаево-Черкесской Республики является: p.W1282* (28 аллелей/70%). Число больных с синдромом псевдо-Барттера составило 54,5%. В 90 % случаев, у пациентов гомо- и гетерозиготных по p.W1282*, отмечалась тяжелая степень панкреатической недостаточности. Заключение:Пациенты с муковисцидозом из Чеченской и Карачаево-Черкесской Республик, несмотря на проведение неонатального скрининга, разработанную и проводимую терапию муковисцидоза, являются сложной категорией пациентов. Отчасти это обусловлено особенностями фенотипа и уникальным распределением аллелей и генотипов гена CFTR. Высокое число гомозиготных вариантов гена CFTR у пациентов из Чеченской и Карачаево-Черкесской Республик, вероятно, обусловлено моноэтнической брачной ассортативностью
Ключевые слова: кистозный фиброз, муковисцидоз, ген CFTR, Чеченская Республика, Карачаево-Черкесская Республика, фенотип, генотип, корреляции, массовое параллельное секвенирование, синдром псевдо-Барттера
Введение. Муковисцидоз (МВ) — это аутосомно-рецессивное наследственное заболевание, возникающее в результате наличия патогенных нуклеотидных вариантов в гене, кодирующем регулятор трансмембранного транспорта ионов хлора (CFTR - Cystic Fibrosis Transmembrane conductance Regulator). Ген CFTR расположен в хромосомной области 7q31.2 и кодирует белок с молекулярной массой 168138 дальтон [1]. Тяжесть течения болезни обусловлена полиорганностью поражения, в структуре которой ведущее место занимают патологические процессы в респираторном тракте. Дыхательная недостаточность и осложнения со стороны органов дыхания определяют большую часть смертельных исходов при МВ [2, 3]. Механизм возникновения и развития МВ хорошо изучен и заключается в нарушении функционирования ответственного за ионный транспорт хлорного канала, синтез которого осуществляется геном CFTR.
В настоящее время описано более 45 тысяч различных полиморфных генетических вариантов гена CFTR согласно ресурсу dbSNP [4]. При этом в базе данных HGMD описано 1673 патогенных варианта, вызывающих МВ. Ряд исследователей выделяют VII классов патогенных нуклеотидных вариантов. К классу VII относят варианты, в результате которых нарушено образование иРНК (информационной РНК). Это могут быть структурные варианты гена CFTR (делеции, инсерции), охватывающие несколько экзонов и нарушающие нормальную структуру гена и нормальный сплайсинг (примером является распространенная в России делеция CFTRdele2,3), либо варианты, изменяющие донорный или акцепторный сайты сплайсинга одного экзона (например, 1717-1G>A) [5, 6]. Однако большинство специалистов считают нецелесообразным выделение этих генетических вариантов из состава I класса. С момента открытия гена CFTR научное сообщество интересует взаимосвязь патогенных вариантов гена с фенотипическими проявлениями заболевания. Показано, что варианты I–III классов негативно влияют на функцию поджелудочной железы, вызывая раннюю панкреатическую недостаточность. Напротив, варианты IV–VI классов обладают протективным эффектом на панкреатическую функцию. Поэтому варианты I–III классов называют «тяжелыми», а IV–VI – «мягкими». При их сочетании в генотипе эффект «мягкого» варианта преобладает над «тяжелым» [7, 8].
Первые исследования взаимосвязи генотипа и фенотипа при МВ были сосредоточены на изучении пациентов с наиболее частым патогенным вариантом p.F508del, который обнаруживается примерно в 70% хромосом МВ в западных странах и обнаруженный в 98 (39,2%) из 250 проанализированных аллелей гена CFTR у российский детей [9]. Castellani и соавт. [7] показали, что гомозиготы p.F508del почти всегда имеют недостаточность поджелудочной железы, тогда как 28% гетерозигот p.F508del и 64% пациентов, которые не являются носителями варианта p.F508del, имеют сохранную функцию поджелудочной железы.
В настоящий момент генотип-фенотипические корреляции у пациентов с МВ из Чеченской и Карачаево-Черкесской Республик не описаны. Между тем заболевание у этих пациентов протекает с особенностями и зачастую имеет плохой прогноз, что вызывает большой научный и практический интерес для изучения клинических и молекулярно-генетических особенности у детей с МВ из Чеченской и Карачаево-Черкесской Республик.
Цель исследования. Изучить клинические и молекулярно-генетические особенности детей с МВ из Чеченской и Карачаево-Черкесской Республик.
Материалы и методы исследования. В исследование включено 237 пациентов, госпитализированных в пульмонологическое отделение ФГАУ «НМИЦ здоровья детей» Минздрава РФ с 2012 по 2022 годы в возрасте от 3 месяцев до 17 лет (средний возраст 8,4±4,6, медиана возраста 7,2) с клинической картиной МВ. Дети были разделены на группы согласно этнической принадлежности: жители Чеченской Республики (ЧР) (61 пациент), жители Карачаево-Черкесской республики (КЧР) (20 человек), пациенты из других субъектов Российской Федерации – 156 детей (ЦФО – 115, ЮФО – 11, ДВФО – 5, УФО – 10, СФО – 5, Приволжский федеральный округ – 10).
Тяжелая степень панкреатической недостаточности диагностировалась в случае снижения содержания панкреатической эластазы в кале менее 50 мкг/г. Поражение печени в виде цирроза определялось с помощью аппарата Фиброскан-502 (метод фиброэластометрии, шкала METAVIR F0-F4). Наличие цилиндрических бронхоэктазов и полипозного пансинусита изучалось с использованием мультиспирального компьютерного томографа Discovery CT750 HD (General Electric) с применением следующих параметров: 100-120 кВт, 80-100 мА, толщина среза 0,625 мм, pitch-1. Спирометрию проводили на аппарате Master Screen (Cardinal Health) в соответствии с рекомендациями Американского торакального и Европейского респираторного обществ по общим принципам определения функциональных показателей легких и стандартизации спирометрии. Выделение геномной ДНК осуществлялось с помощью набора реактивов DNA Blood Mini Kit (QIAGEN, Германия) на автоматизированной станции для выделения нуклеиновых кислот QIAcube (QIAGEN, Германия). Определение концентрации геномной ДНК производилось при помощи флуориметра Qubit 3.0 (Invitrogen, США) с использованием набора Qubit dsDNA HS Kit (Invitrogen, США). Поиск нуклеотидных вариантов осуществлялся методом массового параллельного секвенирования гибридизационной таргетной панели, включающей в себя всю последовательность гена CFTR, в том числе некодирующие области и интроны. Общий размер панели составлял 300 000 нуклеотидов. Секвенирование осуществлялось на платформе MiSeq System (Illumina, США). По результатам секвенирования средняя глубина чтения была 200х, и 99% целевого участка имело покрытие более чем 50х. Все найденные минорные варианты с частотой встречаемости менее 1% согласно базе данных gnomAD (v. 2.1.1), а также нуклеотидные замены, неописанные в базах данных HGMD и dbSNP, были подвергнуты биоинформатическому анализу с использованием программного обеспечения Alamut Visual (Interactive Biosoftware, Франция) на предмет возможной патогенности. Все каузальные нуклеотидные варианты были валидированы при помощи секвенирования методом Сэнгера на генетическом анализаторе 3500xL Genetic Analyzer (Applied Biosystems, США). Все выявленные варианты гена CFTR были описаны согласно номенклатуре HGVS с учетом принятых рекомендаций.
Статистическую обработку данных проводили в среде программирования R с использованием базовых статистических пакетов. Для определения нормальности распределения использовался тест Шапиро-Уилка. Для сравнения групп был использован критерий Манна-Уитни (для количественных переменных), поскольку данные не были нормально распределены. Для качественных переменных применяли точный тест Фишера. Расчет показателей отношение шансов (ОШ) и доверительный интервал (ДИ) производился автоматически при расчете статистических тестов. Степень достоверности определялась на уровне значимости р≤ 0,05 (р – критерий – уровень значимости).
Результаты и их обсуждение. Нами проведен клинический и молекулярно-генетический анализ детей с МВ из ЧР (n=61, из них 33 гомозиготы по p.Y515*). Мутантный аллель p.Y515* выявлен у всех пациентов из ЧР. Количество больных с мекониальным илеусом составило 3/61 (4,91%), преимущественно у гомозигот p.Y515* (10,7% из данной группы). Количество больных с синдромом псевдо-Барттера составило 42/61(68,8%), у 10/61 (16,3%) отсутствовали клинические и лабораторные данные за синдром электролитных нарушений (вероятно, на фоне профилактического выпаивания глюкозо-солевыми растворами), у 9/61 (14,7%) данных об электролитных нарушениях не было. Проявление синдрома псевдо-Барттера у гомо- и гетерозигот p.Y515* составило 23/33 (69,7%) и 19/28 (67,8%) соответственно. Бронхоэктазы выявлены у 26/61 (42,6%) пациентов, преимущественно у гомозигот p.Y515* 18/33 (54,5 %). Полипозный риносинусит выявлен у 36/61 (59%) пациентов. Панкреатическая недостаточность чаще встречается среди гомозигот p.Y515*, 33/33 (100%) в сравнении с гетерозиготами. Результаты положительного неонатального скрининга получены у 21/61 (34,4%) пациентов. Вероятно, это связано с низкой диагностической эффективностью программы неонатального скрининга у детей, проживающих в различных республиках Северного Кавказа [10]. Наличие фиброза и дальнейший переход в цирроз печени выявлены у 3/61 (4,91%) пациентов.
При проведении клинического и молекулярно-генетического анализа детей с МВ из КЧР (n=20, из них 10 гомозиготы по p.W1282*) мутантный аллель p.W1282* выявлен у 19/20 пациентов из КЧР, кроме одного пациента имевшим генотип p.F508del/p.S1231Pfs*4. Количество больных с мекониальным илеусом составило 2/20 (10%); оба пациента были гомозиготными по p.W1282*. Количество больных с синдромом псевдо-Барттера составило 12/20(60%), у 3/20 (15%) отсутствовали клинические и лабораторные данные за синдром электролитных нарушений, у 5/20 (25%) данных об электролитных нарушениях не было. Количество пациентов с проявлением синдрома псевдо-Барттера у гомо- и гетерозигот W1282* составило 7/10 (70%) и 5/9 (55%) соответственно. Бронхоэктазы выявлены у 11/20 (55%) пациентов. Значимых отличий в формировании бронхоэктазов у гомо- и гетерозигот не установлено. Полипозный риносинусит выявлен у 11/20 (55%) пациентов. В 95 % (19/20) случаев, у пациентов гомо- и гетерозиготных по p.W1282* отмечалась тяжелая степень панкреатической недостаточности. Пациент с генотипом p.F508del/p.S1159F имел сохранную функцию поджелудочной железы. У единственного пациента с фиброзом печени (F2 по шкале METAVIR) была обнаружена гомозигота по p.W1282*.
По всей группе исследуемых пациентов с МВ (n=237) было отмечено, что панкреатическая недостаточность выявлена у 210 обследованных детей (84%). Поражение печени в виде цирроза диагностировано у 11 детей (4,4%), при этом у 2-х пациентов проведена пересадка печени от родственного донора. Наличие бронхоэктазов определялось у 150 пациентов (60%), а хронического полипозного риносинусита – у 155 (62%). Проявления синдрома Псевдо-Барттера в виде потери электролитов (калия, натрия, хлора) на первом году жизни установлено у 66 пациентов (26,4%).
Нами также описано распределение спектра частот аллелей и генотипов гена CFTR у пациентов с МВ из Чеченской и Карачаево-Черкесской Республик (Табл. 1, 2). Наиболее частыми аминокислотными вариантами гена CFTR у пациентов с МВ из ЧР являются p.Y515* (94 аллеля /78,3%) и p.E92K (18 аллелей/15%). Наиболее частыми аминокислотными вариантами гена CFTR у пациентов с МВ из ЧКР являются p.W1282* (28 аллелей/70%) и p.F508del (3 аллеля/7,5%)

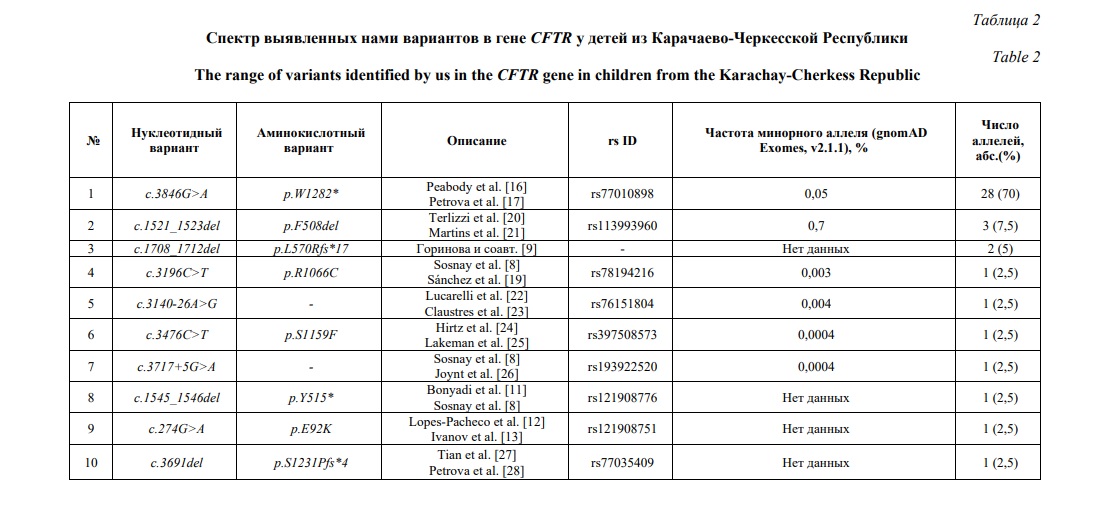
В результате работы у российских детей с МВ из Чеченской и Карачаево-Черкесской Республик выявлены следующие гено-фенотипические особенности. У пациентов из ЧР мекониевый илеус встречается значимо реже, чем среди пациентов с патогенным вариантом p.F508del. Панкреатическая недостаточность встречается чаще среди пациентов гомозиготных по p.Y515* в сравнении с гетерозиготами. В 100 % случаев, у пациентов гомозиготных по p.Y515* отмечалась тяжелая степень панкреатической недостаточности. Гетерозиготное состояние (преимущественно с наличием генетического варианта p.E92K), в свою очередь, чаще является благоприятным прогностическим фактором отсутствия поражения поджелудочной железы, однако отмечается постепенное снижение уровня панкреатической эластазы-1 к более старшему возрасту. Потеря электролитов выявлялась чаще среди пациентов из ЧР, по сравнению с пациентами из других регионов РФ. Количество больных с синдромом псевдо-Барттера в ЧР составило более 60%, при этом у всех пациентов болезнь манифестирует преимущественно с синдрома псевдо-Барттера.
Было установлено, что потеря электролитов выявлялась достоверно чаще среди пациентов из ЧР по сравнению с общей группой пациентов: p <0,001, ОШ 10,79 [95% ДИ 4,64-27,67]. Мекониевый илеус значимо реже встречался среди пациентов с патогенным вариантом p.Y515*, чем среди пациентов с p.F508del: p <0,001, ОШ 0,27 [95% ДИ 0,04-1,05]. Среди детей из ЧР панкреатическая недостаточность достоверно чаще встречается среди пациентов гомозиготных по p.Y515* в сравнении с гетерозиготами. Так, у всех 33 пациентов гомозиготных по p.Y515* выявлена тяжелая степень панкреатической недостаточности. В связи с чем показатель ОШ и 95%-ный доверительный интервал не определены: p <0,001;[95% ДИ 15,28-]. Наблюдается тенденция к постепенному снижению уровня панкреатической эластазы-1 к более старшему возрасту у гетерозигот по p.Y515*. Показано, что в группе пациентов из ЧР бронхоэктазы диагностировались достоверно реже, чем у пациентов из других регионов: p <0,001; ОШ 0,43 [95% ДИ 0,22-0,85], а также при сравнении только с теми, чей генотип включал в себя патогенный вариант p.F508del: p <0,001; ОШ 0,42 [95% ДИ 0,20-0,89]. Достоверных различий по частоте встречаемости полипозного риносинусита и показателям флоуметрии (ОФВ-1 и ФЖЕЛ) среди изученных групп пациентов не установлено.
Среди пациентов из ЧР панкреатическая недостаточность достоверно чаще встречается у пациентов гомозиготных по генетическому варианту p.Y515* в сравнении с пациентами гетерозиготными по p.Y515* и подтверждает высказывание Castellani и соавт. [7] о том, что эффект «мягкого» варианта гена CFTR преобладает над «тяжелым».
У пациентов из КЧР мекониевый илеус встречается крайне редко. Проявление илеуса не зависит от генотипа. Синдром псевдо-Барттера встречается у половины пациентов, преимущественно у гомозигот. Практически у всех пациентов (более 90%) гомо- и гетерозиготных по p.W1282* отмечалась тяжелая степень панкреатической недостаточности.
В ФГАУ «НМИЦ здоровья детей» Минздрава России наблюдается значительное число детей с МВ из различных республик Северного Кавказа. Мы сосредоточились на больных из Чеченской и Карачаево-Черкесской Республик. Согласно регистру пациентов с муковисцидозом в Российской Федерации (РФ) от 2021 года [29], абсолютное число пациентов с данным заболеванием в Чеченской Республике (ЧР) составляет 61, двое из которых- взрослые. В Карачаево-Черкесской Республике (КЧР) 23, один из которых- взрослый. Проанализировав данные регистра с 2015 по 2021 гг [29-35] можно судить о том, что число впервые выявленных пациентов с МВ из Чечни и КЧР увеличивается с каждым годом (Рис. 1).
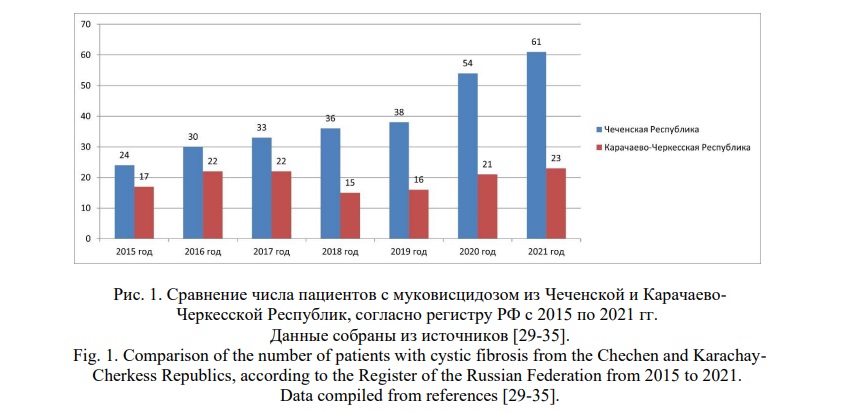
При сравнении наблюдаемых нами пациентов, показан сверхстремительный рост больных из ЧР. Количество больных с 2015 по 2021 гг., увеличилось более чем в два раза, количество больных из КЧР – в 1,3 раза. Первые попытки охарактеризовать особенности генотипа у ЧР проводились как в российской, так и иностранной литературе. Так было показано, что распределение патогенных вариантов гена CFTR в чеченском этносе уникально в отношении высокой частоты двух нуклеотидных вариантов p.Y515* и p.E92K. Показана ассоциация варианта p.E92K с менее тяжелым течением болезни, по сравнению с гомозиготным вариантом p.Y515* [9, 14]. Похожие исследования проводились Петровой Н.В. и соавт. [36], в результате которых был получен спектр аминокислотных вариантов в гене CFTR у представителей основных этнических групп, проживающих на территории КЧР: карачаевцев, ногайцев, черкесов и абазин. У 10 пациентов на 18 из 20 аллелей (90 %) выявлен вариант p.W1282*: восемь пациентов оказались гомозиготами, двое – компаунд-гетерозиготами. В двух последних семьях для возможности идентификации второго аллеля проведено секвенирование кодирующей области гена CFTR и идентифицированы вторые мутантные аллели: p.R1066C – в одном случае и p.Arg709* – в другом. Таким образом, исследование показало, что патогенный вариант p.W1282*, который встречается в разных регионах мира, но наибольшая ее частота выявлена у евреев-ашкеназов Израиля (до 50 % мутантных аллелей у больных с МВ), характерен и для пациентов с МВ из КЧР.
По обобщенной информации о генотипе пациентов, проживающих в республиках Северного Кавказа было показано, что в ЧР генотипированы 96,3% пациентов, а наиболее частый в европейской популяции вариант p.F508del не был обнаружен ни у одного пациента. В КЧР генотипированы 100% пациентов, вариант p.F508del обнаружен у 4,9% пациентов [37]. Что противоречит описанию Петровой Н.В. и соавт. [17] о том, что у карачаевцев, больных МВ, наиболее распространенной в РФ генетический вариант p.F508del в гене CFTR, не обнаружен.
Мажаров В.Н. и соавт. [37], при комплексном изучении эпидемиологии, генетики, клиники и состояния здоровья пациентов, страдающих МВ из семи регионов Северо-Кавказского федерального округа, отмечают, что генетическое исследование у детей с МВ выполнено лишь в 91,7 % случаев (в РФ в целом – в 93,7% случаев). Тогда как в нашем исследовании, генетическое исследование выполнено в 100 % случаев, и у всех исследуемых пациентов были идентифицированы биаллельные каузальные варианты в гене CFTR, в том числе благодаря выбору оптимального метода диагностики.
В ходе анализа состояния здоровья российских пациентов с МВ и динамики основных клинико-лабораторных показателей за 2011–2021 гг [38] было установлено, что к 2021 году наиболее частыми аминокислотными вариантами гена CFTR в РФ являются p.F508del (51,55%), p.CFTRdele2,3 (6,11%), p.E92K (3,46%), p.Y515* (2,25%). Стоить обратить внимание, что в 2011 году частота наиболее частого патогенного варианта p.Y515* для пациентов из ЧР составила меньше 1,06.
Анализ полученных нами данных о распределении частот аллелей и генотипов гена CFTR позволил подтвердить, что патогенные варианты p.Y515* и p.E92K характерны для пациентов из ЧР и составляют 78,3% и 15% мутантных аллелей соответственно, как было описано ранее при исследовании корреляции клинической картины и генотипа у всех обследуемых нами российских детей с МВ [9]. В своих наблюдениях T. Tkemaladze и соавт. [39] также показывают, что аминокислотный вариант p.Y515* является одним из распространенных в Кавказском регионе. Однако в своем исследовании авторы описывают пациентов с МВ, проживающих в западной части Закавказья с генотипом p.Y515*/L997F, клинический фенотип которого может быть непредсказуемым. Пациенты с данным генотипом могут иметь как классический фенотип заболевания, так и обладать не классическими проявлениями МВ, в виде повышения иммунореактивного трипсина, нормальной прибавки массы тела, отсутствия панкреатической недостаточности и поражения бронхолегочной системы в раннем возрасте. В настоящее время данная комбинация аллелей у детей из Чеченской и Карачаево-Черкесской Республик не описана как в нашем исследовании, так и в регистре пациентов с муковисцидозом Северо-Кавказского федерального округа [37]. Обращает на себя внимание и наблюдение того, что описанный нами ранее патогенный вариант p.L570Rfs*17 [9], который выявлен у 2-х пациентов, до сих пор не был описан в международной базе данных [40], но отмечен в Российском регистре МВ [29].
В ходе оценки исходов мекониевого илеуса [41] было доказано, что впоследствии у пациентов с непроходимостью кишечника отмечалась склонность к формированию синдрома псевдо-Барттера (11 из 138 больных), но в указанных патогенных вариантах, при которых отмечался илеус, не включен вариант p.Y515*. Таким образом, учитывая полученные данные у пациентов из ЧР, можно судить о том, что корреляции между генотипом, включавшем вариант p.Y515* и появлением мекониального илеуса и в дальнейшем, как следствие, проявление синдрома псевдо-Барттера у детей с МВ из ЧР, выявлено не было. Нами показано, что потеря электролитов выявлялась чаще среди пациентов из ЧР (в сравнении с общей группой пациентов, имевших генотип, включавший вариант p.F508del, так и без него).
Синдром псевдо-Барттера неоднократно описан как в российских, так и в зарубежных исследованиях [42], нередко этот синдром диагностируется как «истинный» синдром Барттера [43]. Основное отличие синдрома псевдо-Барттера от истинного синдрома заключается в отсутствии потери натрия с мочой и нарушений в почечных канальцах. Так, Mantoo M.R. и соавт. [44] описал ключевые различия между данными синдромами. Впервые признаки, схожие с синдромом потери электролитов у детей с МВ, описывал в 1951 году Kessler W.R и Andersen D.H [45]. В дальнейшем синдром нередко описывался у детей с МВ, особенно у пациентов, проживающих в местности с жарким климатом [46, 47]. Так, у пациентов из ЧР, вне зависимости от погодных условий, отмечается преимущественное начало заболевания с синдрома псевдо-Барттера. Внутри чеченского этноса, достоверных различий в проявлении синдрома потери электролитов у гомо- и гетерозигот по p.Y515* выявлено не было. Такая же тенденция отмечается и у пациентов из КЧР, то есть проявления электролитных нарушений, вне зависимости от погодных условий. Исходя из того, что патогенные варианты p.Y515*, p.E92K и p.W1282* характерны для пациентов из ЧР и КЧР соответственно, данные результаты позволяют оптимизировать медико-генетическое консультирование в данных регионах РФ.
По результатам нашего исследования установлены аллельные частоты вариантов гена CFTR у жителей ЧР (Табл. 1). Мажаров В.Н и соавт. [37] указывают схожие относительные частоты выявленных вариантов в гене CFTR у пациентов из ЧР. Однако обращает на себя внимание тот факт, что в регистре не указано 2 патогенных варианта p.D110H и p.L145Ffs*8, которые выявлены у наблюдаемых нами пациентов, что может свидетельствовать о неполноте заявленного ранее регистра.
В настоящее время, наиболее частый для европейской и российской популяций патогенный вариант p.F508del не был обнаружен ни у одного пациента из ЧР. У пациентов из КЧР напротив, доля p.F508del составила 7,5% мутантных аллелей, что также как и результаты Мажарова В.Н и соавт. [37] противоречат высказыванию Петровой Н.В. и соавт. [36] о том, что у карачаевцев, больных МВ, не обнаружен мутантный аллель p.F508del. Петрова НВ и соавт [48] также изучали особенности спектра и частот патогенных вариантов гена CFTR в популяциях Северного Кавказа. Проанализировав результаты генетического тестирования 165 неродственных пациентов с МВ из Северо-Кавказского федерального округа, были получены высокие доли вариантов p.W1282* (72,0%) и p.Y515* (69,8%) соответственно. Частота варианта p.W1282* у карачаевцев составила 88,9%, доля p.F508del составила 6%.
Проанализировав клинические и молекулярно-генетические особенности муковисцидоза у детей из ЧР и КЧР, а также преимущественное начало заболевания с синдрома псевдо-Барттера у пациентов из ЧР и наличие электролитных нарушений у половины пациентов из КЧР, можно достоверно констатировать, что данным пациентам необходима оценка электролитов в крови, а также профилактическое выпаивание глюкозо–солевыми растворами для выявления и соответственно снижения риска возникновения электролитных нарушений.
Заключение. Пациенты с МВ из Чеченской и Карачаево-Черкесской Республик, несмотря на проведение неонатального скрининга, разработанную и проводимую терапию МВ, являются сложной категорией пациентов. Отчасти это обусловлено распределением аллелей и генотипов гена CFTR у пациентов с МВ из ЧР: оно уникально в отношении высокой частоты патогенных вариантов p.Y515* и p.E92K, в тоже время вариант p.F508del не был обнаружен ни у одного пациента. Основной особенностью данных пациентов, является преимущественное начало заболевания с синдрома псевдо-Барттера. Наличие в генотипе патогенного варианта p.E92K связано с менее тяжелым течением болезни, чем у гомозигот по p.Y515*, в виде сохранной функции поджелудочной железы в детском возрасте. Патогенный вариант p.W1282* характерен для пациентов из КЧР, встречается у 95% представителей этого этноса и составляет 70% мутантных аллелей. Доля варианта p.F508del составила 7,5 %. У пациентов, обладающих вариантом p.W1282* в гомо- и гетерозиготном состоянии, более чем в 90% случаев, отмечалась тяжелая степень панкреатической недостаточности. Высокое число гомозиготных вариантов гена CFTR у пациентов из Чеченской и Карачаево-Черкесской Республик, вероятно, обусловлено моноэтнической брачной ассортативностью.
Информация о финансировании
Работа выполнена в рамках государственного задания «Изучение этиологических особенностей редких болезней, имеющих патогенетическую терапию» № 1220032300501-0.














Список литературы