
Этиология и патогенез атеросклероза: современные представления и возрастзависимые аспекты (обзор)
Aннотация
Актуальность: Атеросклероз является ведущей причиной заболеваемости и смертности населения во всём мире. Он представлет собой системный патологический процесс, выражающийся в отложении атерогенных липопротеинов в интиме сосудов, с дальнейшим развитием воспаления, клеточной пролиферации и образованием атеросклеротических бляшек, приводящий к обтурации просвета сосуда. Несмотря на накопленный к текущему моменту довольно значительный материал по этиологии и патогенезу атеросклероза современные взляды на механизмы и эволюцию этого патологического процесса, его возрастзависимые аспекты требуют обобщения и систематизации. Данное обстоятельство послужило основанимем для анализа данных мировой литературы, посвященной вопросам этиологии и патогенеза атеросклероза и особенностей его фомирования в возрастзависимом контексте. Цель исследования:Рассмотреть с современных позиций вклад основных этиологических факторов в развитие атеросклероза и обобщить их значимость для формирования данной патологии на системном, органном, тканевом, клеточном и молекулярном уровнях в возрастзависимом контексте. Материалы и методы:Проведён анализ литературных источников и баз данных Web Of Science, ScienceDirect, Medline, Российского РИНЦ, Google Scholar, PubMed, Semantic Scholar, Taylor & Francis, Wiley Online Library и Bielefeld Academic Search Engine по проблеме этиопатогенеза атеросклероза, опубликованных за последние 10 лет. Результаты:В данном обзоре суммированы современные данные о факторах риска и механизмах развития атеросклероза (возраст, пол, курение, микробиота, воспаление, эндотелиальная дисфункция, роль в этом процессе регуляторных РНК). С современных позиций рассмотрена эволюция атеросклеротической бляшки и возрастзависимые аспекты болезни. Дана детальная характеристика роли некодирующих РНК в развитии и эволюции атеросклеротических бляшек. Заключение:Атеросклероз представляет собой сложный патологический процесс, в формировании которого играет роль большое число факторов, как генетических, так и средовых, взамодействие которых определяет характер и темпы прогрессирования заболевания
Ключевые слова: атеросклероз, этиология, патогенез, факторы риска, эндотелиальная дисфункция, регуляторные РНК
Введение. В структуре смертности населения большинства развитых и развивающихся стран мира сердечно-сосудистые заболевания занимают лидирующие позиции на протяжении многих десятилетий. По данным ВОЗ в России пациентов с болезнями системы кровообращения насчитываются около 31 млн. человек [1]. Наиболее распространённой причиной таких форм патологии является атеросклероз. Это системный патологический процесс, выражающийся в отложении атерогенных липопротеинов в интиме сосудов, с дальнейшим развитием воспаления, клеточной пролиферации и образованием атеросклеротических бляшек и обтурации просвета сосуда. Данные процессы заканчиваются прогрессирующей ишемией органов и тканей с появлением в них склеротических и некротических изменений [2, 3]. В частности, атеросклероз является патологическим процессом, который лежит в основе развития неблагоприятных сосудистых событий, включая ишемическую болезнь сердца (ИБС), инсульт и поражение периферических артерий, ответственных за большую часть сердечно-сосудистой заболеваемости и смертности в современном мире [3, 4, 5]. Эпидемиологические исследования показывают, что распространенность атеросклероза увеличивается во всем мире и, вероятно, достигнет масштабов эпидемии в ближайшие десятилетия [1, 2].
Общепринято, что атеросклероз представляет собой заболевание, характеризующееся накоплением липидов, фиброзных элементов и кальцификацией в крупных артериях. В основе этого процесса лежит активация эндотелия, за которой следует каскад событий, предполагающий вазоконстрикцию и активацию воспалительных механизмов, приводящих в итоге к образованию атероматозных бляшек [5-8]. Поскольку определить ведущий этиологический фактор развития атеросклероза на сегодняшний день по-прежнему не представляется возможным, выделяют целый ряд наиболее значимых факторов риска его развития. В современной литературе достаточно хорошо детально освещены вопросы характирестики модифицируемых и немодифицируемых факторов риска развития атеросклероза. Среди них выделяют пол и возраст, курение, гиподинамия, неправильное питание, дислипидемия, гипергликемия, артериальная гипертензия, ожирение [9-12]. Многочисленные исследования показывают, что старение является важнейшим фактором, определяющим темпы и характерразвития атеросклеротического процесса, тем самым, способствуя преждевременному или ускоренному морфологическому изменению (старению) сосудов.
Несмотря на накопленный к текущему моменту довольно значительный материал по этиологии и патогенезу атеросклероза современные взляды на механизмы и эволюцию этого патологического процесса, его возрастзависимые аспекты требуют обобщения и систематизации. Данное обстоятельство послужило основанимем для анализа данных мировой литературы за последние 10 лет, посвященной вопросам этиологии и патогенеза атеросклероза и особенностей его фомирования в возрастзависимом контексте.
Цель исследования. Рассмотреть с современных позиций вклад основных этиологических факторов в развитие атеросклероза и обобщить их значимость для формирования данной патологии на системном, органном, тканевом, клеточном и молекулярном уровнях в возрастзависимом котексте.
Материалы и методы исследования. Для достижения поставленной цели был проведён анализ литературных источников по проблеме этиопатогенеза атеросклероза, опубликованных за последние 10 лет. Поиск научной информации проводили в базах данных Scopus (http://scopus.com/), Web Of Science (https://www.webofscience.com/), ScienceDirect (https://www.sciencedirect.com/), Medline (https://medlineplus.gov/), Российском индексе научного цитирования (РИНЦ, https://elibrary.ru/), а также в поисковых системах Google Scholar (https://scholar.google.com/), PubMed (https://pubmed.ncbi.nlm.nih.gov/), Semantic Scholar (https://www.semanticscholar.org/), Taylor & Francis (https://www.tandfonline.com/), Wiley Online Library (https://onlinelibrary.wiley.com/) и Bielefeld Academic Search Engine (BASE, https://www.base-search.net/).
Результаты и их обсуждение
1. Возраст и пол как важнейшие факторы риска развития атеросклероза
На сегодняшний день принято считать, что особенности в закономерностях развития атеросклероза у женщин и мужчин обусловлены врожденными биологическими и социальными различиями [11, 13]. Ещё в первой половине прошлого столетия в США было выполнено Фремингемское исследование, которое показало, что половой диморфизм в основных модифицируемых факторах риска, включая курение сигарет, дислипидемию, гипертонию и сахарный диабет, может быть причиной наблюдаемых различий в развитии и/или осложнениях атеросклероза [13]. У женщин по сравнению с мужчинами выше риск развития инфаркта миокарда (ИМ) с отношением шансов 1.3, 1.5 и 1.6 для курения, гипертонии и сахарного диабета 2 типа соответственно, что было показано на крупных выборках пациентов [14]. Кроме того, более молодые женщины имеют меньший риск развития сердечно-сосудистых заболеваний и более низкую частоту развития ИМ по сравнению с мужчинами, о чём свидетельствуют данные современных эпидемиологических исследований [15, 16]. Однако, в возрасте от 60 до 79 лет у женщин исчезает отмечавшийся кардиопротекторный эффект. Риск же сердечно-сосудистых заболеваний (ССЗ) у женщин превышает таковой у мужчин к 80 годам [16]. Тем не менее, эта модель начала заболевания не повторяется при инсульте. Этот факт может свидетельствовать о большей распространенности у женщин до 70 лет высокой вероятности развития повторного инсульта в течение первых 5 лет после инсульта по сравнению с мужчинами [17]. Кроме того, ишемическая болезнь сердца (ИБС) часто не диагностируется и рассматривается как ведущая причина женской смертности. Так, например, в Европе на ССЗ приходится 43% смертей у мужчин и 55% у женщин [13]. При анализе различных компонентов ССЗ на ишемическую болезнь сердца (ИБС) приходится 21% смертей у мужчин и 23% у женщин, при этом, как указывалось выше, инсульт является более частой причиной смерти у женщин, чем у мужчин (18% и 11% соответственно), а также другие ССЗ (15% у женщин и 11% у мужчин). Эти демографические статистические данные свидетельствуют о половых различиях в риске сердечно-сосудистых заболеваний и подчеркивают необходимость учитывать пол как важную переменную [17].
Несмотря на то, что в последнее время были предприняты значительные усилия для понимания молекулярных механизмов и открытия новых мишеней для лекарств в терапии атеросклероза, понимание природы полового диморфизма подверженности атеросклерозу все еще относительно ограничено. Масштабное исследование, в котором была проанализирована 771 доклиническая статья об атеросклерозе и других сосудистых заболеваниях, показало, что пол животных не указан в 18,8% из них. При указании пола 55,4% исследований проводились на самцах, 20,4% - на самках и менее 25% - как на самцах, так и на самках [13, 18]. Доля исследований, включающих оба пола, была по некоторым данным очень похожей (21–28%) [18]. Менее половины исследований на людях, включающих оба пола, напрямую сравнивали показатели мужчин и женщин, рассматривая пол как независимую переменную или его связь с генотипом и эффективностью лечения в контексте наличия атеросклероза [18].
В ряде исследований показано, что ишемия напряжения (например, стенокардия и перемежающаяся хромота) нередко возникает, когда отдельные бляшки увеличиваются настолько, что ухудшают кровоток для удовлетворения потребностей тканей (обычно стеноз >70%) [19]. С другой стороны, разрыв бляшки приводит к наибольшей части случаев заболеваемости и смертности от атеросклероза, таких как инфаркт миокарда, инсульт, инвалидизирующее заболевание периферических артерий и, в конечном итоге, смерть [20]. В этом отношении особое место отводится неинвазивным методам диагностики. Так, ультразвуковая визуализация показала, что у мужчин бляшки развиваются раньше и имеют большее количество, чем у женщин, даже с учетом различий в факторах риска. Кроме того, бляшки со стенозом 30–40% чаще разрываются, что приводит к окклюзии сосудов и смерти, что показали посмертные патологоанатомические исследования [19]. Эти данные убедительно показывают, что общее количество бляшек, воспалительное состояние бляшек и нестабильная морфология бляшек способствуют развитию острого ИМ и риска инсульта. Это также согласуется с большей частотой ишемических событий у мужчин, хотя со временем эта взаимосвязь может меняться [20]. Кроме того, имеются данные, что у женщин не так много атеросклеротических бляшек, как у мужчин, и у них меньше признаков бляшек высокого риска [21]. При этом, у мужчин наблюдалось значительно больше атеросклеротических и кальцифицированных бляшек, более высокая частота ИБС и серьезных неблагоприятных сердечных событий в течение 5-6 лет наблюдения у мужчин по сравнению с женщинами [13]. Кроме того, кросс-секционное исследование REFINE-Рейкъявик (включало 21132 пациента) показало, что у 50% женщин результаты компьютерной томографии были нормальными, по сравнению с 31% мужчин [13].
Новые методы визуализации позволяют оценить индивидуальные особенности атеросклеротических бляшек, связанные с неблагоприятными событиями. Внутрисосудистое ультразвуковое исследование, которое дает представление, как о степени стеноза, так и о степени некротического ядра в бляшке, показало, что у женщин, страдающих острым коронарным синдромом, одинаковое количество критических поражений по сравнению с мужчинами [20]. Тем не менее, у женщин наблюдается меньше критических поражений, меньше вовлеченных коронарных артерий с поражениями, более низкая частота разрыва бляшки и меньший общий объем некротического ядра несмотря на более высокий средний возраст и множественные сопутствующие заболевания [21].
Важность понимания возрастных и гендерных различий в составе критических бляшек может иметь решающее значение для выбора более подходящей фармакологической и интервенционной терапии [22]. Так, значительно более высокая распространенность тонкослойной фиброатеромы у пациентов пожилого возраста по сравнению с мужчинами свидетельствует о возможной важности интенсивной гиполипидемической терапии для женщин [22, 23].
В отличие от лечения и профилактики уязвимых бляшек, терапевтическая стратегия при кальцификации коронарных артерий может быть более сложной и трудной. Для интервенционного лечения поражений с большой кальцификацией интракоронарные методы визуализации могут обеспечить более безопасные процедуры и лучшие клинические результаты [24]. Тем не менее, при поражениях с большими бляшками важно использовать устройства для уменьшения объема, а последующая более широкая площадь стента также используется для кальцификации [13, 25]. Кроме того, изменение образа жизни может быть более важным для женщин, чем для мужчин, в отношении снижения риска атеросклеротических изменений в компонентах бляшки и последующего развития заболевания, поскольку они считаются модифицируемыми факторами [13, 26].
2. Курение сигарет как ведущий средовой фактор риска развития атеросклероза
Курение сигарет является важным самостоятельным фактором риска развития атеросклероза и сердечно-сосудистых заболеваний, поскольку химические компоненты дыма обладают высокими окислительными и воспалительными свойствами, которые могут непосредственно вызывать повреждение эндотелия и усиливать воспалительную реакцию [27]. Клинические данные показали прямо пропорциональную дозозависимую связь воздействия курения с наличием обширных и кальцифицированных атеросклеротических бляшек, а отказ от курения в любом возрасте является одной из наиболее важных медицинских рекомендаций для снижения риска атеросклеротических сердечно-сосудистых заболеваний, рака и смертности [28]. Отказ от курения был связан с меньшим прогрессированием каротидной бляшки, но не с увеличением толщины интимы-медиа [29]. Влияние отказа от курения на атеросклероз сонных артерий зависело от степени воздержания и сохранялось после поправки на исходную тяжесть курения и факторы риска атеросклеротических сердечно-сосудистых заболеваний [29].
В работе J. Gambardella и соавт. было показано, что отдельные вещества табакокурения (например, никотин, карбонильные соединения, акролеин и метилвинилкетон) и/или их комбинированное действие могут влиять на каждую стадию атеросклеротического процесса в отдельности [27]. Воздействие дыма может способствовать развитию окислительного стресса. Это состояние является одним из основных механизмов, лежащих в основе повреждения эндотелия. Доказано, что окислительный стресс влияет на активность многих ферментов (например, eNOS и НАДФН-оксидаз) и приводит к необратимой модификации различных белков, существенно изменяя тем самым внутриклеточные сигнальные пути [13]. Также, прямая окислительная активность компонентов дыма способствует метаболическим изменениям и индукции окисления липопротеидов низкой плотности (ЛПНП), что может повышать их уровень [27]. Известно, что воздействие дыма также увеличивает экспрессию молекул адгезии на плазматической мембране и активирует воспалительные гены, включая ИЛ-1 и ЦОГ-2 [13]. Это, в свою очередь, приводит к активации NF-κB в эндотелиальных клетках (ЭК) [18]. Кроме того, никотин может запускать секрецию провоспалительных адипокинов из околососудистой жировой ткани [30]. Более того, курение индуцирует пролиферацию и миграцию гладкомышечных клеток сосудов (ГМКС), а, также, их переключение с сократительного на секреторный фенотип вследствие увеличения экспрессии IFN-β и PDGF [31]. Следовательно, ГМКС способны высвобождать провоспалительные факторы и компоненты внеклеточного матрикса.
Атеросклеротические бляшки курильщиков характеризуются преобладанием липидного ядра, а фиброзная «шапочка» тоньше, чем у некурящих [32]. Эта конфигурация частично обусловлена повышенной активностью матриксных металлопротеиназ (ММП) в бляшках курильщиков [13]. Курение запускает инфильтрацию и активацию макрофагов внутри очага поражения, а также их превращение в пенистые клетки, способствуя росту липидного ядра. Курение также играет значимую роль в процессе активации тромбоцитов и их адгезии к эндотелию [13]. В дополнение к этим эффектам курение может способствовать повышению артериального давления, что является определяющим фактором повреждения и нестабильности атеросклеротических бляшек.
По некоторым данным, есть убедительные результаты, позволяющие подтвердить факт воздействия сигаретного дыма на регуляцию некоторых путей микроРНК, участвующих в развитии атеросклероза [33]. Так, было показано, что воздействие высоких доз сигаретного дыма приводит к усилению микроРНК-155 (miR-155) и микрРНК-21 (miR-21), нацеленных на активируемые пероксисомными пролифераторами рецепторы – PPAR-α. Понижение уровня PPAR-α приводит к усилению молекул адгезии (VCAM-1, ICAM-1) и хемокинов (MCP-1) за счет активации транскрипционного фактора AP-1 [13, 34]. При этом, VCAM-1 и ICAM-1 опосредуют прочную адгезию лейкоцитов к ЭК и играют критическую роль в последующей миграции лейкоцитов, что приводит к развитию атеросклероза [35]. Экспрессия MCP-1, в свою очередь, может регулировать миграцию и инфильтрацию моноцитов/макрофагов [18]. Кроме того, активация miR-155 и miR-21 может модулировать в ЭК пути EGFR/ERK/p38 MAPK и PI3K/Akt/eNOS соответственно [13, 36].
3. Роль микробиоты в развитии атеросклероза
В человеческом теле обитает множество видов бактерий (микробиота), которые проходили путь эволюции вместе с людьми, и создали симбиотические отношения, способные приносить пользу друг другу. Микробиота способна положительно влиять на физиологические процессы в организме человека, принимая участие в синтезе различных метаболитов или, например, стимуляции иммунной системы [37]. Вместе с тем, влияние микроорганизмов также было может быть связано с развитием различных нозологий, таких как атеросклероз или сердечно-сосудистые заболевания (ССЗ) [37, 38].
По данным ряда исследований присутствие микроорганизмов в атероматозной бляшке хорошо известно, а также, установлены корреляционные связи между наличием большого числа определенных типов микробов и стабильностью бляшки наряду с общей воспалительной реакцией [39]. Например, было показано, что бактериальная ДНК из патогенных семейств, таких как Helicobacteraceae или Neisseriaceae, более распространена в бляшках пациентов с симптомами атеросклеротического поражения [40].
В последнее время всё большее число работ указывает на участие микробиоты кишечника в развитии атеросклероза и сердечно-сосудистых заболеваний. Человеческий кишечник содержит самую большую микробную популяцию и способен регулировать многие биологические функции, такие как накопление энергии, поглощение и переработка питательных веществ или созревание иммунной системы [41]. Однако, когда микробный гомеостаз кишечника нарушается в сторону недостаточно представленных колоний, т.е. развивается дисбактериоз, то это может вызывать развитие различных заболеваний, таких как сахарный диабет 2 типа, ожирение, онкологический процесс, сердечно-сосудистые заболевания и атеросклероз [41]. Кроме того, дисбиоз кишечника увеличивает его проницаемость, снижая экспрессию белков плотных контактов и делая возможной транслокацию липополисахаридов, вызывающих вялотекущее воспаление через Toll-подобные рецепторы [42]. С другой стороны, некоторые метаболиты, продуцируемые микробиотой кишечника, могут модулировать системное воспаление и способствовать или предотвращать развитие атеросклероза. Наиболее изученным является триметиламин-N-оксид (ТМАО) [42], окисленная форма триметиламина (ТМА). ТМА синтезируется микробиотой кишечника после метаболизма диетического холина и карнитина. Затем он переносится в кровоток и окисляется до ТМАО в печени с помощью флавинмонооксигеназы [43]. ТМАО усиливает воспалительные реакции сосудистой стенки и подавляет обратный транспорт холестерина, способствуя накоплению холестерина в интиме [43]. Кроме того, у мышей APOE-/-, получавших пищу, богатую холином, наблюдались повышенные уровни ТМАО и развитие атеросклеротических бляшек, этот процесс прекращался при введении антибиотиков [13, 42].
Непрямые инфекции в отдаленных от атеромы участках также способствуют ее развитию и дестабилизации. Действительно, в последние годы многие исследования связывают частоту атеросклеротических сердечно-сосудистых заболеваний с бактериальными инфекциями. Так, подтверждение получила идея причинно-следственной связи между заболеваниями пародонта или хламидийными инфекциями и сердечно-сосудистыми заболеваниями в ряде эпидемиологических исследований [13, 44]. Большое исследование, в котором приняли участие почти 12 000 человек, показало, что плохая гигиена полости рта связана с повышенным риском сердечно-сосудистых заболеваний и усилением менее выраженного воспаления [13, 44]. Кроме того, небольшое исследование с 92 участниками показало, что некоторые типы бактерий были более многочисленны в ротовой полости пациентов с симптоматическим атеросклерозом, чем у пациентов контрольной группы, что указывает на возможную связь между бактериями полости рта и развитием атеросклеротических бляшек [44]. Тем не менее, влияние бактерий полости рта на развитие атеросклеротических сердечно-сосудистых заболеваний все еще остается спорной [13, 45].
4. Воспаление как патологический процесс, связанный с развитием атеросклероза
Как известно, воспалительные процессы вовлечены во все фазы развития атеросклероза [46]. На ранних стадиях атеросклероза ЛПНП накапливаются и модифицируются в субэндотелиальной области. Известно, что под воздействем модифицированных ЛПНП гладкомышечные клетки сосудов высвобождают хемоаттрактанты, в том числе хемокины 2 (CCL2) и CCL5 [13]. Эти факторы могут способствовать привлечению моноцитов [46]. Кроме того, окисленные ЛПНП (оксЛПНП) и минимально модифицированные ЛПНП (ммЛПНП) способны индуцировать провоспалительную реакцию в ЭК и макрофагах, усиливать повреждение эндотелия и привлекать лейкоциты. Также, ммЛПНП могут связываться с TLR2 и классом 4 PRR и через путь NF-kB индуцировать секрецию провоспалительных цитокинов IL-1β, IL-6 и TNF-α [13, 46].
По некоторым данным CD36-опосредованное поглощение оксЛПНП активирует инфламмасому NLRP3. Такая активация приводит к усиленной секреции провоспалительного цитокина ИЛ-1β36 [47]. Кроме того, воспалительные реакции в макрофагах и дендритных клетках способны вызывать иммунные комплексы оксЛПНП со специфическими антителами. Эти иммунные комплексы индуцируют клеточную активацию, продукцию воспалительных цитокинов и образование пенистых клеток. В этом случае главный путь передачи сигналов осуществляется через рецептор Fc гамма-рецептора I иммунного комплекса (FcγRI) [47]. Также, они могут активировать инфламмасомы посредством FcγR/TLR4/CD36-зависимого механизма [13, 46]. На ранних стадиях атеросклероза высвобождение CCL2 и Т-клеточных хемоаттрактантов привлекает моноциты и лимфоциты во внутреннюю артериальную стенку, где моноциты дифференцируются в пенистые клетки макрофагов под влиянием M-CSF [13, 48]. Миграцию и пролиферацию ГМКС при образовании жировой полоски индуцируют Т-клетки, секретируя TNF-β, IFN-γ, фиброгенные медиаторы и факторы роста. Стимуляция продукции ММР макрофагами в очаге поражения также происходит посредством активации Т-лимфоцитов [47].
В настоящее время имеется свидетельство того, что посредством повреждения лизосом кристаллы холестерина могут активировать инфламмасому NLRP3 [13, 47]. В ряде исследований установлено, что активированные эндотелиальные клетки чрезмерно экспрессируют молекулы клеточной адгезии, такие как VCAM-1, ICAM-1, PECAM-1, E-селектини и P-селектин, на своей поверхности [13, 46]. Таким образом, эти активированные ЭК являются локальным источником привлечения лейкоцитов в участки атеросклеротического поражения [46]. Существует мнение, что ЭК, макрофаги и ГМКС экспрессируют LOX-1, который связывается с оксЛПНП, что приводит к увеличению продукции свободных радикалов кислорода и гибели ГМКС. Это способствует инфильтрации оксЛПНП в эндотелий, что нарушает нормальную функцию эндотелия, адгезию моноцитов и инфильтрацию [49]. Известно, что простагландин E2 (PGЕ2) является важным липидным медиатором-эйкозаноидом, который опосредует такие патофизиологические явления, как лихорадка, боль и воспаление. PGE2 синтезируется из арахидоновой кислоты при участии фермента циклооксигеназы (ЦОГ), а затем и PGE2-синтазы (PGES). Было показано, что истощение микросомальных PGES в миелоидных клетках, но не в ЭК или ГМКС, способствует атерогенезу у мышей [18]. Кроме того, имеются данные, что рецепторы простагландинов связаны с ранним атеросклерозом и воспалительным процессом, вызывающим эрозию и разрыв бляшки [13, 46].
5. Дисфункция эндотелия и его связь с развитием атеросклероза
Эндотелий сосудов представляет собой гетерогенный монослой, образованный эндотелиальными клетками (ЭК). Эти клетки представляют собой первый барьер для молекул, клеток или патогенов, циркулирующих в кровотоке, поскольку обеспечивают просвет всех кровеносных сосудов [13, 46]. В крупных сосудах стенки выстланы одним слоем ЭК, называемым эндотелием, который вместе с коллагеновыми и эластическими волокнами образует просветный слой сосудов или интиму. При этом, ЭК находятся в тесном контакте со средней оболочкой, состоящей из гладкомышечных клеток сосудов (ГМКС), а также эластической и коллагеновой ткани. Кроме того, этот слой окружает адвентициальная оболочка, состоящая в основном из плотного матрикса соединительной ткани. С другой стороны, стенки артериол и венул состоят из тех же трех слоев, что и более крупные сосуды, хотя медии и адвентиции гораздо тоньше и менее выражены. Наконец, посткапиллярные венулы полностью лишены медии и адвентиции и состоят только из ЭК и базальной мембраны [13].
Эндотелий расположен между циркулирующей кровью и тканями. Он работает как датчик и преобразователь сигналов, синтезируя биологически активные вещества. Все изменения в циркулирующей крови воспринимаются эндотелием, который затем опосредует передачу сигнала на другие слои сосудистой стенки. К таким изменениям относятся механические напряжения (удлинение и напряжение сдвига стенки – WSS от англ. wall shear stress), а также изменения концентрации метаболических факторов [13]. Некоторые физиологические функции, такие как регуляция гомеостаза, сосудистого тонуса и целостности сосудов могут модулироваться различными механическими силами, действующие на артериальную стенку. Известно, что помимо их роли в гомеостазе, участие гемодинамики в развитии сосудистых заболеваний имеет решающее значение в патологии атеросклероза, влияя как на начало заболевания, так и на его прогрессирование. Основные силы, действующие на артериальную стенку, включают как растягивающее напряжение, вызванное кровяным давлением, так и WSS – тангенциальную силу к стенке сосуда, вызванную кровотоком, которая играет важную роль в атерогенной гемодинамике [50]. Сегменты сосудов с низким WSS или сильно колебательным WSS, по-видимому, подвергаются наибольшему риску развития атеросклероза [46]. Изменение WSS может непосредственно влиять на морфологию и функцию эндотелия сосудов и стимулировать миграцию и пролиферацию ГМКС и мононуклеарных клеток [13, 46]. Низкое или нестабильное изменение WSS является индикатором для оценки гемодинамических изменений, которые тесно связаны с атеросклерозом [13, 48]. Величина и направление WSS распознаются механосенсорами эндотелия и передаются как биохимические сигналы. Индуцированная WSS механотрансдукция регулирует экспрессию многочисленных генов, участвующих в клеточной морфологии, адгезии и пролиферации [13, 48]. Например, напряжение сдвига вызывает характерное выравнивание ЭК [51]. В трубчатых или прямых участках артерий, где WSS представляет собой ламинарный поток, ЭК имеют уплощенную форму и удлиненное выравнивание в направлении потока [13]. Однако в местах бифуркации или высокой кривизны сосуда возникает нарушение потока и, как следствие турбулентного и реверсивного потока с пониженным WSS на внешней стенке сосуда, ЭК увеличивают свой объем, принимая вид булыжника [50]. Более того, гемодинамические силы определяют раннее развитие локализованных атеросклеротических бляшек, которые не распределены случайным образом ни в экспериментальных животных моделях, ни у людей [50, 52]. Атеросклеротические поражения в основном возникают в областях, характеризующихся низкой WSS и отрывом потока, и чаще всего включают точки ветвления и бифуркации. Обширные корреляционные данные, указывающие на то, что низкий сдвиг или нарушенный поток объясняют локализацию атеросклеротических поражений, подчеркивают важность артериальных ветвей и бифуркаций при постановке диагноза развития или прогрессирования атеросклеротического поражения [13].
Эндотелий модулирует тонус подлежащих гладких мышц сосудов; поддерживает неадгезивную поверхность просвета; и опосредует гемостаз, клеточную пролиферацию, воспалительный и иммунный ответ в сосудистой стенке [13, 52]. Фактически эндотелий высвобождает как агонисты, так и антагонисты, чтобы сбалансировать эффект в обоих направлениях. Например, ЭК способны продуцировать как коагулянты или антикоагулянты, вазодилататоры или вазоконстрикторы, так и провоспалительные или противовоспалительные молекулы [13, 52].
Согласно современным представлениям, атеросклероз инициируется эндотелиальной дисфункцией, сопровождающейся задержкой липопротеидов низкой плотности (ЛПНП) и их модификацией в интиме [53]. Модифицированные ЛПНП вместе с дополнительными атерогенными факторами способствуют активации ЭК, что приводит к привлечению моноцитов в интиму. Модифицированные ЛПНП активно захватываются дифференцированными моноцитами и ГМКС, что способствует образованию пенистых клеток [53]. Кроме того, активируются несколько воспалительных сигнальных путей, что приводит к образованию жировых полос, что является первым признаком атеросклероза и характеризуется значительным накоплением липидов как внутри клеток (макрофаги и ГМКС), так и во внеклеточном пространстве [13]. Известно, что нарушение механизмов, участвующих в регуляции сосудистого гомеостаза, приводит к дисфункции эндотелия [52, 54]. Иными словами, когда ЭК теряют способность поддерживать гомеостаз, стенки сосудов предрасположены к вазоконстрикции, инфильтрации липидов, адгезии лейкоцитов, и, кроме того, активации тромбоцитов и окислительному стрессу, среди прочего [54]. Вместе они вызывают воспалительную реакцию, которая считается первым этапом образования атероматозной бляшки: жировая полоса [52, 54]. Кроме того, эндотелиальная дисфункция также играет заметную роль в последующих стадиях атеросклероза, участвуя в развитии бляшки и ее разрыве на последних стадиях атеросклероза [54]. В этой связи повышенная эндотелиальная дисфункция считается ранним индикатором атерогенеза [55].
В последнее время локальным фактором риска атерогенеза, способствующим эндотелиальной дисфункции, часто называют гемодинамические силы [56]. Как указано выше, участки, подверженные поражению, в основном расположены в областях, где ламинарный поток нарушен из-за расслоения потока, рециркуляции или повторного прикрепления [57]. Этот турбулентный поток создает временные и пространственные градиенты, что приводит к более высокому колебательному индексу и более низкому напряжению сдвига [57]. Кроме того, нарушенный поток также способствует инфильтрации липопротеинов в интиму сосуда, во-первых, потому, что ЛПНП остаются в этих областях в течение более длительных периодов времени, а во-вторых, потому что турбулентный поток вызывает физическое нарушение целостности эндотелия, что способствует инфильтрации липопротеинов [57]. Кроме того, еще одна фундаментальная связь между гемодинамическими силами и атерогенезом связана с экспрессией различных эндотелиальных генов, регулируемых кровяными механическими стимулами [58]. Влияние напряжения сдвига на экспрессию эндотелиальных генов изучалось в течение последних 20 лет; к настоящему времени обнаружено более 40 генов, участвующих в этом процессе [58]. Среди них несколько атерогенных генов, таких как хемоаттрактантный белок моноцитов 1 (MCP-1), который индуцирует проникновение моноцитов в артериальную стенку [13], и тромбоцитарные факторы роста (PDGF), которые усиливают миграцию ГМКС и активируются в ЭК [58]. Интересно, что данные исследований выявили элементы реакции на сдвиг (SSRE) в промоторах этих и других генов, таких как eNOS или молекула адгезии тромбоцитов-1 (PECAM-1), которые способствуют развитию бляшек [13]. Более того, комбинация двух или более SSRE в одном и том же промоторе может иметь синергетический эффект, усиливающий экспрессию этих генов [58]. С другой стороны, в прямолинейных участках сосудистой сети, где ламинарный поток вызывает высокое напряжение сдвига в эндотелии, некоторые проатерогенные гены подавляются, в то время как гены, вызывающие остановку клеточного цикла или увеличивающие антиоксидантную способность, активируются. Действительно, длительное воздействие на ЭК ненарушенного ламинарного потока способствует усилению эндотелиальной синтазы оксида азота (eNOS), тем самым увеличивая их способность к синтезу оксида азота (NO) [52]. Эти данные свидетельствуют о дифференцированном молекулярном ответе эндотелия в зависимости от паттерна кровотока, подчеркивая роль гемодинамических сил в эндотелиальной дисфункции.
Эндотелиальную дисфункцию также принято объяснять снижением биодоступности NO [57]. NO синтезируется из L-аргинина в ЭК в ходе реакции, катализируемой eNOS, и диффундирует через клеточные мембраны, достигая гладкомышечной ткани стенки артерии. NO способствует релаксации гладких мышечных волокон, известной как эндотелийзависимая вазодилатация, и считается атерозащитной молекулой, поскольку противодействует атерогенезу и его осложнениям [52]. В частности, NO участвует в снижении агрегации тромбоцитов, окислении тканей и воспалении, а также активации тромбогенных факторов; и влияет на рост, пролиферацию и миграцию клеток [59] (Рис. 1). Более того, он поддерживает метаболический гомеостаз, так как снижает содержание триглицеридов и стеатоз, увеличивает синтез инсулина, клиренс глюкозы и эффективность митохондрий [60]. Однако при наличии сердечно-сосудистых факторов риска, таких как гиперлипидемия, гипертензия, курение или диабет, продукция NO снижается вследствие повышенного окислительного стресса, который обычно связан с этими патологиями [57, 61]. Окислительный стресс способствует синтезу проатерогенных цитокинов (TNF-α и интерлейкинов IL-1 и IL-6), молекул адгезии (VCAM-I и ICAM-I) и хемокинов (MCP-1) посредством активации NF-kB белками теплового шока (HSP-60). Эти медиаторы ингибируют активность eNOS и, следовательно, продукцию NO [52] (Рис. 1). Фактически, исследования, проведенные у пациентов с гиперхолестеринемией, продемонстрировали нарушение эндотелийзависимой вазодилатации из-за дефекта биодоступности NO [13, 52]. У пациентов с артериальной гипертензией также обнаруживается дефект эндотелиальной NO-системы, что может объяснять нарушение эндотелий-зависимой вазодилатации при сердчено-сосудистой патологии [13, 61].
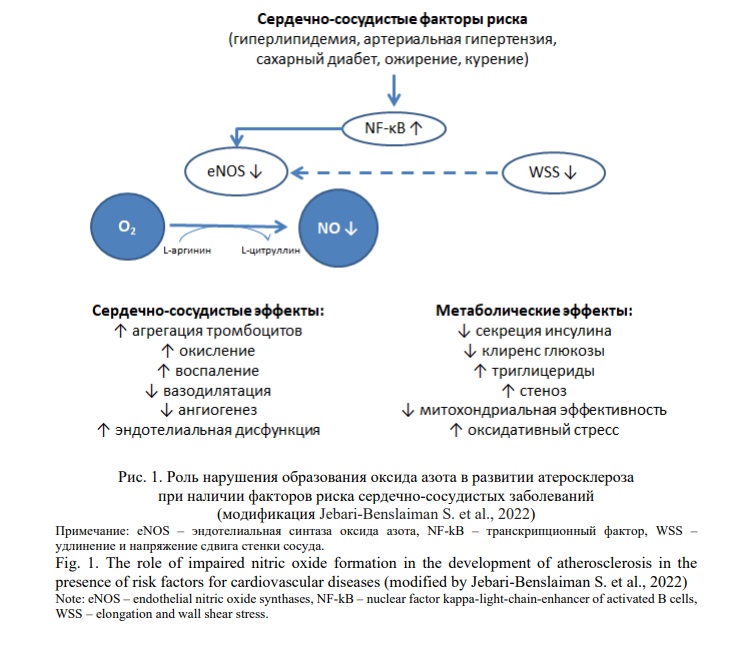
6. Сосудистая инфильтрация липопротеидов и формирование жировой полоски
Накопление липопротеидов низкой плотности (ЛПНП) в плазме способствует трансэндотелиальной инфильтрации циркулирующих ЛПНП в интиму. Хотя традиционно считалось, что ЛПНП проходят эндотелий путем диффузии или парацеллюлярно [53], в настоящее время признано, что трансцитоз играет важную роль в трансэндотелиальном транспорте ЛПНП [62]. В частности, было показано, что трансцитоз ЛПНП опосредуется рецептором-мусорщиком B1 (SR-B1) и рецептором, подобным рецептору активина А типа 1 (ALK1) эндотелия, что отличается от классического пути эндоцитоза ЛПНП, опосредованного LDLR [62]. Рецепторы SR-B1 и ALK1 локализуются совместно с кавеолами, что указывает на то, что трансцитоз ЛПНП с помощью SRB1 и ALK1 опосредуется зависимым от кавеол механизмом [53]. Кроме того, отсутствие кавеолина-1, основного структурного белка кавеол в ЭК, значительно ухудшает транспорт и удержание ЛПНП в артериальной стенке [62], а повышенные уровни кавеолина-1 были обнаружены при атеросклеротических поражениях [63]. Несмотря на то, что для выяснения специфического транспортного механизма обоих рецепторов считают необходимым проведение дополнительных экспериментов, эти данные весьма убедительно свидетельствуют о том, что зависимое от кавеол поглощение ЛПНП играет значимую роль в трансцитозе ЛПНП [64]. Таким образом, обобщая роль ЭК в инфильтрации ЛПНП, важно отметить, что следует учитывать и другие факторы, такие как гликокаликс, перициты, субэндотелиальный внеклеточный матрикс и роль напряжения сдвига [13, 65].
Оказавшись в субэндотелиальном пространстве, захваченные частицы ЛПНП окисляются, чему способствует отсутствие защитных антиоксидантов плазмы, таких как токоферол, аскорбат, ураты, аполипопротеины или сывороточный альбумин [64]. Окисленные ЛПНП являются ключевыми воспалительными компонентами, которые способствуют развитию атеросклеротических бляшек, поскольку они содержат окисленные липиды и продукты их деградации, принимающие активное участие в механизмах развития заболевания [13, 62]. ЛПНП окисляются свободными радикалами, присутствующими во внеклеточной среде, такими как супероксид (O2•–), гидроксильные радикалы (•OH) и другие, такие как HClO, продуцируемые окружающими клетками [61]. Кроме того, ЛПНП могут быть непосредственно окислены ферментативной активностью фосфолипаз и липоксигеназ [57]. Таким образом, липоксигеназный путь был выделен для объяснения инициации окисления ЛПНП. Интересно, что белок, связанный с рецептором ЛПНП (LRP), участвует в привлечении ЛПНП, а липоксигеназа 12/15 перемещается к мембране, где происходит окисление эфиров холестерина ЛПНП [13, 57].
Независимо от механизма, вовлеченного в инициирование окисления ЛПНП, этот процесс сопровождается снижением уровня антиоксидантов, включая альфа-токоферол и каротиноиды [13]. Далее происходит деградация полиненасыщенных жирных кислот (ПНЖК), в основном арахидоновой и линолевой кислот, которые окисляются до гидропероксидов. Последнее приводит к образованию сопряженных диенов и при дальнейшем окислении короткоцепочечных альдегидов [13, 61]. АпоВ-100, основной белок ЛПНП, также подвергается различным модификациям вследствие воздействия окислительной среды. Например, альдегиды, образующиеся при окислении липидов, образуют соединения с лизиновыми остатками апоВ-100. Вместо этого апоВ-100 может быть непосредственно модифицирован преимущественно по остаткам тирозина с помощью окисляющих агентов [61]. Эти модификации ингибируют распознавание ЛПНП соответствующими рецепторами, тем самым увеличивая захват частиц липопротеинов через нерегулируемые рецепторы [13, 61].
В зависимости от уровня окисления окисленные ЛПНП классифицируются как минимально модифицированные ЛПНП (ммЛНП) или экстенсивно окисленные ЛПНП (оксЛПНП) [61]. Мм-ЛПНП химически отличаются от немодифицированных ЛПНП, но все же распознаются рецепторами-ЛПНП и, следовательно, интернализируются через регулируемые пути. Однако модифицированные липиды внутри этих частиц действуют как биологически активные молекулы, придающие другие биологические активности, не проявляющиеся в немодифицированных ЛПНП [61]. Эти биоактивные липиды могут вызывать провоспалительный ответ в ЭК и макрофагах [66]. С другой стороны, когда ЛПНП сильно модифицированы, они становятся неузнаваемыми рецепторами-ЛПНП, в то же время позволяя распознавать ряд рецепторов-мусорщиков [67]. Окислительные модификации апоВ-100 лежат в основе отсутствия сродства к рецепторам-ЛПНП и повышения сродства к рецепторам-мусорщикам. Более того, оксЛПНП способны ускользать от задержки протеогликанов, что способствует их нерегулируемому поглощению рецепторами-мусорщиками [66]. После интернализации продукты, полученные из оксЛПНП, запускают экспрессию молекул воспаления в макрофагах, о чём отдельно будет изложено ниже. Необходимо также отметить, что, хотя окисление ЛПНП является наиболее распространенной модификацией, ряд модификаций ЛПНП, способствующих развитию атеросклероза, таких как гликозилирование, ацетилирование и агрегация также описан в ряде других работ [68].
Особое значение в развитии атеросклероза играет стимуляция эндотелия, также известная как активация эндотелия I типа, происходит, когда воспалительные агенты вызывают изменение тонуса микрососудов, их проницаемости или диапедеза лейкоцитов [67]. Это явление представляет собой острую реакцию с кратковременными функциональными и морфологическими изменениями и не требует синтеза белка de novo или активации генов [68]. Однако в ответ на некоторые провоспалительные агенты, такие как ИЛ-1, ФНО, эндотоксины, модифицированные липопротеины и конечные продукты гликозилирования (AGE), а также на биомеханическую стимуляцию, вызванную нарушением кровотока, эндотелий может подвергаться устойчивой фенотипической модуляции, известной как активация эндотелия II типа [67, 68]. Эта активация приводит к сложному воспалительному ответу, который начинается с увеличения продукции NF-kB в ЭК в ответ на вышеупомянутый стимул. NF-kB повышает экспрессию молекул адгезии лейкоцитов, таких как VCAM-1 и ICAM-1; секретируемые хемокины, такие как MCP-1 и ИЛ-8 [69]; и протромботические медиаторы, такие как ингибитор активатора плазминогена или тканевой фактор. Активированные ЭК индуцируют избирательное привлечение моноцитов в интиму. Этот процесс был визуализирован in vitro [70], и его представляют как последовательную смену роллинга, адгезии, активации и трансмиграции моноцитов, как было отмечено в целом ряде работ [13, 68, 71]. Привлечение моноцитов начинается с захвата и перемещения их по эндотелию, что в основном опосредуется Р-селектином [67, 69]. Роллинг моноцитов затем снижается, и моноциты остаются прочно прикрепленными к эндотелию, что опосредовано связыванием интегринов моноцитов с VCAM-I и ICAM-I ЭК [13, 70]. Кроме того, при качении по эндотелию моноциты активируются связанными с поверхностью эндотелия хемокинами, такими как CXCL1, CXCL2, CXCL4 и CCL5, что увеличивает адгезивность моноцитов [71]. После этого моноциты трансмигрируют в пространство интимы. Это движение включает пересечение барьера ЭК, его базальной мембраны и слоя перицитов [70, 71]. Процесс миграции поддерживается хемокинами, которые ранее секретировались в ответ на провоспалительные сигналы.
В настоящее время наиболее частым хемокином, опосредующим трансмиграцию моноцитов является MCP-1 (также называемый CCL2), однако также было изучено влияние других хемокинов, таких как CCL3, CCL4 и CCL5 [72]. МСР-1 продуцируется в основном эндотелиальными клетками, гладкомышечными клетками, моноцитами и макрофагами интимы, и его экспрессия повышается после провоспалительного стимула или повреждения ткани, способствуя трансэндотелиальной миграции циркулирующих моноцитов из плазмы в интиму [72]. Этот процесс опосредован парацеллюлярным и трансцеллюлярным путями [65]. При парацеллюлярном пути миграция моноцитов происходит преимущественно через соединения ЭК из-за перераспределения соединительных молекул в воспаленном эндотелии [71]. Кроме того, некоторые молекулы эндотелиального соединения активно опосредуют этот тип миграции [68]. С другой стороны, при трансцеллюлярном пути клетки мигрируют через тело ЭК, однако этот тип трансмиграции наблюдался только в 10–30% событий in vitro [64]. Считается, что новые достижения в визуализации живых клеток могут прояснить эту проблему [13, 70]. Наконец, моноциты пересекают базальную мембрану ЭК, которая состоит из сети ламинина и коллагена, и оболочки перицитов, которая обнаруживается в большинстве венул [70]. Оказавшись в интиме, моноциты дифференцируются в макрофаги, которые могут быть поляризованы по фенотипу М1 (провоспалительный) или М2 (противовоспалительный) [73]. Тем не менее, макрофаги проявляют чувствительность к изменениям воспалительной среды и в ответ на новые сигналы способны переключать свой фенотип с провоспалительного на противовоспалительный [73]. Пластичность макрофагов имеет основополагающее значение для успешного ответа с преобладанием М1 при прогрессировании заболевания и М2 при регрессии [73, 74]. Макрофаги M1 высвобождают воспалительные цитокины и хемокины и продуцируют NO и активные формы кислорода (АФК), которые способствуют привлечению моноцитов и дальнейшему распространению воспалительной реакции [74].
Необходимо отметить, что макрофаги также экспрессируют ряд рецепторов, которые опосредуют интернализацию модифицированных и немодифицированных ЛПНП. Как упоминалось ранее, липопротеины, оставшиеся в интиме, подвержены модификациям из-за воспалительной среды, что позволяет им интернализоваться через рецепторы-ловушки CD36, SRA-1 и LOX-I [67]. Важно подчеркнуть, что экспрессия этих рецепторов не подавляется поглощением холестерина. Таким образом, в контексте атеросклероза, когда содержание оксЛПНП значительно повышено, клетки интернализуют большее количество оксЛПНП. Внутри клеток оксЛПНП расщепляются в лизосомах, а холестерин, содержащийся в липопротеинах, этерифицируется ацил-КоА-холестеролацилтрансферазой (АСАТ) в эндоплазматическом ретикулуме (ЭР). Эфиры холестерина хранятся в виде капель липидов, расположенных как в цитоплазме, так и связанных с ЭР [75]. Гидролиз этих упакованных эфиров холестерина, опосредованный гидролазами нейтральных эфиров холестерина, такими как nCEH и NCECH1, приводит к образованию свободного холестерина, который переносится из макрофагов в апоА1 или ЛПВП (липопротеины высокой плотности), что является важным шагом для удаления избытка холестерина из периферических тканей [75]. Этот процесс опосредован АТФ-связывающими кассетными транспортерами ABCA1 и ABCG1 и переносчиками холестерина SR-B1, которые играют важную роль в обеспечении оттока холестерина из клеток и предотвращении образования пенистых клеток [67]. Однако провоспалительная микросреда атеросклеротических поражений нарушает систему оттока ABCA1 как в макрофагах М1, так и в макрофагах М2 и способствует накоплению пенистых клеток, как показано в экспериментах с мышиными макрофагами, способствующими развитию бляшек [13, 67].
Кроме того, избыточное поглощение липидов макрофагами поддерживает воспалительную реакцию, а оксЛПНП индуцируют сигнальные каскады, которые активируют мишени NF-kB, поддерживающие активацию ЭК, привлечение моноцитов и образование пенистых клеток [74, 75]. Поглощение оксЛПНП макрофагами можно рассматривать как защитный механизм, поскольку они удаляют цитотоксические элементы из интимы. Однако повышенная миграция моноцитов в интиму и последующая дифференцировка в макрофаги приводят к большому количеству пенистых клеток, индуцирующих рост атеросклеротического поражения [74]. Поэтому накопление холестерина считается признаком атеросклеротических поражений [67, 75].
Накопление холестерина в субэндотелиальном компартменте также способствует образованию кристаллов холестерина как внутри, так и вне клеток и способствует развитию атеросклеротических бляшек [74, 75]. Этот процесс наблюдали как снаружи, так и внутри клеток, в макрофагах, инкубированных с оксЛПНП [76]. Хотя кристаллы холестерина являются общим признаком прогрессирующих атеросклеротических поражений, они присутствуют также в ранних бляшках и могут использоваться в качестве маркера раннего развития атеросклероза [76]. Кристаллы холестерина в бляшке активируют инфламмасому NLRP3 в макрофагах, что приводит к активации провоспалительных путей. Инфламмасомы представляют собой цитозольные мультипротеиновые комплексы врожденного иммунитета, ответственные за активацию воспалительных путей [77]. Хотя активация и сборка NLRP3 до конца не изучены, известно, что она приводит к запуску каспазы-1. Каспаза-1 впоследствии расщепляет провоспалительное семейство цитокинов ИЛ-1 на их биоактивные формы, ИЛ-1β и ИЛ-18, способствуя воспалению [77]. Было высказано предположение, что поглощение оксЛПНП, опосредованное рецептором CD36, ответственно за активацию NLRP3 [77]. По-видимому, рецептор-мусорщик CD36 вместе с TLR4-TLR6 поглощает оксЛПНП, что приводит к образованию внутриклеточных кристаллов холестерина. Эти кристаллы вызывают дестабилизацию лизосом, индуцируя высвобождение лизосомального содержимого, такого как катепсины или активные формы кислорода, что приводит к сборке инфламмасомы NLRP3 и последующей активации каспазы-1 [77].
В настоящее время считается, что ГМКС, расположенные в интиме, также способны поглощать оксЛПНП нерегулируемым образом через различные рецепторы-мусорщики, такие как SR-A, CD36 и LOX-1 [68, 77]. Действительно, их вклад в общую популяцию пенистых клеток в бляшке значителен [53]. Кроме того, ГМКС интимы экспрессируют меньше транспортеров ABCA1, чем транспортеры средней оболочки [64, 68]. Следовательно, баланс между входом и выходом холестерина смещается в пользу накопления холестерина и образования пенистых клеток. Кроме того, по крайней мере, 50% пенистых клеток в интиме коронарных артерий человека происходят из ГМКС, а не из моноцитов, что определяет значимость роли ГМКС в развитии атеросклероза [13, 64, 68].
7. Эволюция фиброзной сосудистой бляшки
Во время развития фиброзного процесса атеромные бляшки претерпевают переход от жировой полосы к росту интимы, это этап, характеризующийся образованием бесклеточной и богатой липидами области, известной как некротическое ядро. Чтобы стабилизировать бляшку, некротическая сердцевина покрывается волокнами, образуя фиброзную покрышку. Некротическое ядро и фиброзная покрышка являются отличительной чертой прогрессирующего атеросклероза, и считается маловероятным, что на этой стадии может произойти регресс атероматозных бляшек [76].
Фиброзная шапочка (или покрышка бляшки) представляет собой субэндотелиальный барьер между просветом сосуда и атеросклеротическим некротическим ядром, состоящим из ГМКС, мигрировавших на просветную сторону артерии, и внеклеточного матрикса (ВКМ), полученного из ГМКС [78]. Фиброзная покрышка служит структурной опорой, чтобы избежать обнажения протромботического материала ядра, что в случае нарушения её целостности могло бы вызвать тромбоз [78]. В обычных физиологических условиях дифференцированные ГМКС средней оболочки (медии) проявляют сократительный фенотип, который регулирует диаметр кровеносных сосудов и кровоток [76, 78]. Однако, в ответ на повреждение ГМКС меняют свой фенотип на синтетический, в котором преобладают миграционная и пролиферативная активность [79]. С этой целью соседние клетки активируют процесс заживления, вырабатывая несколько факторов роста, в том числе эпидермальный фактор роста, фактор роста фибробластов, инсулиноподобный фактор роста, фактор роста тромбоцитов (PDGF), трансформирующий фактор роста-β (TGF-β) и фактор роста эндотелия сосудов (VEGF) [79]. При развитии атеросклероза в ответ на факторы роста, продуцируемые пенистыми клетками (полученными из ГМКС или макрофагов) или ЭК интимы, ГМКС из медии оболочки мигрируют в интиму [78]. Более того, ИЛ-1, продуцируемый макрофагами, усиливает эндогенную продукцию PDGF ГМКС и, попав в интиму, аутокринно приводит к их пролиферации [79]. В дополнение к миграции и последующей пролиферации ГМКС с синтетическим фенотипом также увеличивают продукцию компонентов ВКМ, таких как интерстициальный коллаген, эластин и протеогликаны [80]. Эти пролиферирующие ГМКС, наряду с продукцией ВКМ, создают фиброзный колпачок, который покрывает развивающуюся атеросклеротическую бляшку, таким образом, окружая поражение и предотвращая её разрыв [80]. Интересно, что, если продукция митогена не прекращается, ГМКС не переключаются обратно на сократительный фенотип [79, 80], что способствует развитию поражения. Характеристики фиброзной покрышки, такие как толщина, клеточность, состав матрикса и содержание коллагена, являются важными факторами, определяющими стабильность бляшки [78].
Некротическое ядро занимает центральную часть атеросклеротических бляшек. Это ядро, покрытое фиброзной оболочкой, состоит из нагруженной липидами гипоцеллюлярной области с редуцированным поддерживающим коллагеном [79]. По мере развития атеросклеротических бляшек некротическое ядро увеличивается в размерах главным образом вследствие двух факторов: гибели макрофагов и нарушения эффероцитоза – процесса, удаляющего апоптотические клетки. Оба события вносят вклад в воспалительное микроокружение, окислительный стресс и тромбогенность, а также способствуют гибели соседних клеток, таких как ГМКС, повышая уязвимость бляшек [81].
На ранних стадиях атеросклероза апоптоз макрофагов программируется через координированную систему каспаз, что приводит к гибели клеток и эффероцитозу [81]. Однако при развитии атеросклероза в макрофагах и ГМКС усиливается апоптоз из-за повышенного окислительного стресса, активации рецепторов, участвующих в передаче сигналов смерти, ингибирования путей выживания и лишения питательных веществ [82]. На этом этапе фагоцитарная активность макрофагов не способна справиться с накоплением апоптотических клеток, которые подвергаются вторичной некротической гибели с сопутствующим высвобождением внутриклеточных окислительных и воспалительных компонентов. Эта ситуация усугубляет воспаление и усиливает гибель соседних клеток [81, 82]. Эффероцитоз также становится дефектным при прогрессирующем атеросклерозе из-за нарушения активности нескольких рецепторов, которые опосредуют этот процесс, таких как MerTK, LRP1, CD36 и SR-B1 [82]. Кроме того, окисленные фосфолипиды и оксЛПНП, присутствующие в прогрессирующих атеросклеротических бляшках, ингибируют распознавание апоптотических клеток эффероцитотическими рецепторами [82]. Нарушение эффероцитоза в запущенных бляшках также способствует накоплению кристаллов холестерина из апоптотических клеток [82]. В физиологических условиях мелкие кристаллы холестерина быстро секвестрируются из интерстициального пространства клетками-фагоцитами; однако по мере прогрессирования поражения фагоцитирующие клетки не могут удалить избыток кристаллов, которые в конечном итоге увеличиваются в размерах и остаются в субэндотелиальном пространстве [83]. Этот процесс активируется комплементом и увеличивает провоспалительное состояние бляшки. Более того, поскольку фагоцитирующие клетки не способны поглощать крупные кристаллы холестерина рецепторами-мусорщиками, их лизосомальное содержимое секретируется непосредственно в интерстициальное пространство, способствуя более интенсивному иммунному ответу [84]. Эти события способствуют накоплению мертвых клеток и росту некротического ядра. Кроме того, металлопротеиназы, высвобождаемые после гибели клеток, уменьшают размер фиброзной покрышки и повышают уязвимость бляшек [13, 84]. Наконец, апоптотические и некротизированные клетки высвобождают тканевой фактор (TF), который вместе с окисленными липидами увеличивает тромбогенность некротического ядра [13, 81].
Обызвествление бляшек атеромы является еще одним признаком запущенного атеросклероза. Он существует в виде костеподобного образования внутри бляшки и инициируется в воспалительных областях с локальным уменьшением коллагеновых волокон [85]. Высвобождение матриксных везикул после гибели макрофагов и синтетических ГМКС инициирует процесс кальцификации бляшки [74]. В местах образования ядра накапливается ортофосфат кальция, который превращается в аморфный фосфат кальция и, наконец, в кристаллические структуры [85]. Кроме того, процессу кальцификации способствуют и другие факторы, в том числе сниженный уровень ингибиторов минерализации или усиление остеогенной трансдифференцировки [85]. В частности, перициты и ГМКС трансдифференцируются в остеобластоподобные фенотипы во время развития атеросклероза, приобретая способность генерировать минерализованный матрикс и приводя к отложениям кальция, как это происходит в костной ткани [13, 78, 85]. В совокупности это способствует микрокальцификации, ранней стадии каскада сосудистой кальцификации как в интиме, так и в медии [85]. Затем микрокальцинаты превращаются в более крупные кальцинаты, которые распространяются от дна некротического ядра до окружающего матрикса [86]. Хотя кальцификация является отличительной чертой прогрессирующего атеросклероза (она очень хорошо коррелирует с размером бляшки), количество и размер отложений кальция не коррелируют с уязвимостью бляшки, которая скорее связана с другими особенностями, такими как расположение, тип кальцификации или окружающая среда [85, 86].
Как отмечено выше, атеросклеротические бляшки обычно развиваются в разветвленных участках, где WSS ниже. В этих областях низкое напряжение сдвига способствует локальной эндотелиальной дисфункции и эксцентрическому накоплению бляшек. Первоначально сужение просвета предотвращается ремоделированием сосудов наружу для поддержания нормального просвета и восстановления распределения напряжения сдвига. Однако это продлевает локальные, неблагоприятные условия с низким WSS и усугубляет эксцентрический рост бляшки. Эксцентрические бляшки в сохраненных местах просвета испытывают повышенную нагрузку растяжением на свои «плечи», превращая атеросклеротическое изменение в сосудистое поражение, склонное к разрыву [85]. Бляшка считается уязвимой, когда такое поражение имеет большое некротическое ядро, тонкую фиброзную покрышку и повышенную воспалительную реакцию из-за постоянного воздействия проатерогенной среды [86]. Фиброзная «шапочка» отделяет тромбогенно-некротическое ядро от циркулирующих факторов свертывания крови и тромбоцитов, а её толщина коррелирует с уязвимостью бляшки [78, 79]. В результате гибели ГМКС продукция ВКМ снижается, а количество высвобождаемых матриксных металлопротеиназ (ММР) увеличивается, что делает фиброзную покрышку более слабой [78, 87].
Как упоминалось ранее, воспаление способствует развитию бляшки на всех стадиях от возникновения до разрыва бляшки. Действительно, на этой последней стадии его значение существенно возрастает, так как оно способствует нестабильности фиброзной покрышки [87]. Некоторые провоспалительные цитокины, такие как IFN-γ, могут ингибировать выработку коллагена в ГМКС. Кроме того, медиаторы воспаления, обычно обнаруживаемые в атероме, такие как IL-1β, TNF-α и лиганд CD40 (CD154), могут повышать экспрессию MMP в ГМКС, что наблюдается in vitro [13, 88]. Эта воспалительная стадия обычно наблюдается в «шапочке» и «плечах» бляшки вместо генерализованного воспаления [81]. В совокупности данные показывают, что, когда преобладает воспаление, сохранение прочной и жесткой фиброзной покрышки снижается, что делает такую покрышку нестабильной и восприимчивой к разрыву при воздействии гемодинамических сил, что является наиболее распространенным механизмом разрыва бляшки [81, 88].
Известно, что в момент, когда бляшка изъязвляется или разрывается, субэндотелиальное пространство подвергается воздействию крови, запуская процесс коагуляции, чтобы покрыть рану [88]. Первоначально тромбоциты прилипают к субэндотелиальному коллагену и активируются, а затем вовлекаются и агрегируются дополнительные тромбоциты в этой области, чтобы инициировать заживление раны [89]. Одновременно высвобождаются протромботические элементы липидного ядра, которые вступают в контакт с факторами коагуляции плазмы. Более конкретно, тканевой фактор липидного ядра реагирует с фактором VII плазмы, активируя каскад гемокоагуляции, который приводит к продукции тромбина, необходимого промежуточного продукта для образования фибрина [81]. Фибрин представляет собой нерастворимый белок, который образует сети фибриновых нитей и вместе с тромбоцитами покрывает очаг поражения, образуя стабильную и хорошо организованную структуру. Это структурное образование известно как тромб [13, 89]. Хотя целью этого процесса является заживление ран, запуск биохимического каскада способствует расширению интимы в просветную сторону сосуда. Например, активированные тромбоциты высвобождают TGF-β, который, как указывалось ранее, способствует выработке интерстициального коллагена и, следовательно, утолщению фиброзной покрышки [89]. Следовательно, атеросклеротическое поражение расширяется, что приводит к сужению просвета. Все это предполагает отсутствие клинических осложнений.
Развитие тромба запускает ряд реакций, которые делают поражение более фиброзным и стабильным и, следовательно, менее склонным к разрыву. Однако из-за роста бляшек возрастает риск закупорки сосудов. Следовательно, кровоток в коронарных артериях снижается, вызывая ишемические проявления, такие, например, как ишемическая болезнь сердца (стенокардия) и, впоследствии, сердечная недостаточность [87]. Более того, если обтурация полная или почти полная, это приводит к инфаркту миокарда или инсульту [88]. При отделении тромба от артериальной стенки образуется сгусток, известный как эмбол, который способен циркулировать в сердечно-сосудистой системе. В конце концов, эмбол может остановиться в дистальных артериях, где он препятствует кровотоку, что приводит к локальной ишемии, дисфункции органов или потенциальному инфаркту [87]. Однако, если воспалительная реакция прекращается вовремя, например, из-за гиполипидемической терапии, может формироваться стабильная бляшка с достаточным просветом для правильного кровообращения [86].
8. Влияние регуляторных РНК на эволюцию атеросклеротических бляшек
По данным ряда исследований показано, что искаженная экспрессия и функция некодирующей РНК (нкРНК) вовлечены в развитие атеросклеротического процесса. В частности, микроРНК (миРНК) и длинные некодирующие РНК (днРНК) были определены как важные регуляторы развития атеросклеротических бляшек [13, 90, 91]. В то время как миРНК, как известно, регулируют экспрессию генов посттранскрипционно, в основном посредством деградации мРНК, днРНК-опосредованная регуляция менее известна, в основном из-за ее низкой консервативности последовательности и низкой экспрессии [90]. Однако днРНК может активировать и репрессировать гены с помощью различных механизмов, как на уровне транскрипции, так и на уровне трансляции, и ее значение в развитии атеросклероза было впоследствии обновлено [13, 91]. В настоящее время признано, что они играют решающую роль в развитии атеросклероза [90, 91].
На сегодняшний день не вызывает сомнений тот факт, что микроРНК (miR) являются важными молекулами для поддержания сердечно-сосудистого гомеостаза [90]. Действительно, многие исследования последних лет показали, что нарушение регуляции экспрессии микроРНК влияет на клеточные и молекулярные процессы, которые способствуют атеросклерозу [92, 93]. Патологическая основа образования и развития атеросклеротических бляшек связана с уровнями экспрессии соответствующих микроРНК (Табл. 1). В атеросклеротически измененных коронарных артериях выявлены повышенные уровни miR-29, miR-100, miR-155, miR-199, miR-221, miR-363, miR-497, miR-508 и miR-181; напротив, было обнаружено, что miR-1273, miR-490, miR-24 и miR-1284 подавляются [92, 93]. В случае локализации атеросклероза в зонах аорты, бедренных и сонных артерий в атеросклеротических бляшках обнаруживаются повышенные уровни miR-21, miR-34, miR-146 и miR-210 [92, 94]. Кроме того, исследования, проведенные в каротидных бляшках, показали активацию miR-15, miR-26, miR-30, miR-98, miR-125, miR-152, miR-181, miR-100, miR-127, miR- 133, miR-145 и miR-422 и подавление miR-520 и miR-105 [94].
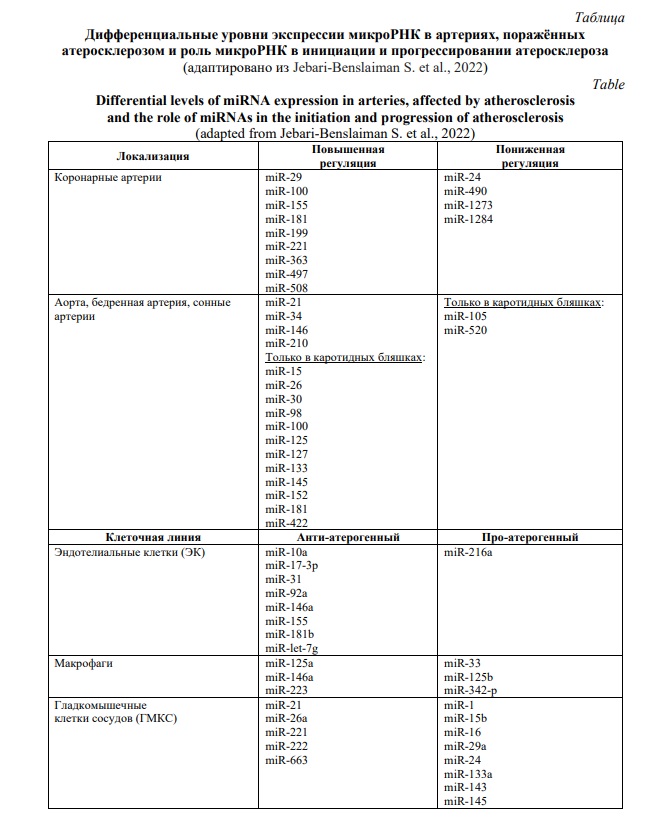
Первые этапы в развитии атеросклероза включают эндотелиальную дисфункцию, за которой следует воспалительная реакция и образование пенистых клеток. Существует обширный список миРНК, регулирующих функцию эндотелия, таких как miR-221, miR-503, miR-217, miR-34a, miR-181b, miR-155, miR-126, miR-1, miR-223, miR-145, miR-146a, miR-92a или miR10a (Табл. 1). Так, известно, что экспрессия miR-155 индуцирует подавление эндотелина-1 и рецептора ангиотензина II типа I, указывая на роль в защите эндотелия [95]. Более того, miR-10a проявляет атеропротективную роль за счет репрессии передачи сигналов GATA6/VCAM1 внутри ЭК [13, 94]. Известно, что miR-10a, miR-31 и miR-17-3p регулируют воспаление, модулируя экспрессию молекул адгезии в ЭК [13, 92]. Кроме того, miR-126, одна из наиболее изученных миРНК, играет роль в профилактике атеросклероза, регулируя путь метаболизма фактора роста эндотелия сосудов (VEGF) и ингибируя проницаемость эндотелия [13, 92]. Другим регулятором эндотелиального воспалительного ответа является miR-181b, который регулирует путь NF-kB [13, 92]. MiR-146a не только снижает поглощение липидов макрофагами, что указывает на атеропротективную роль, но также ингибирует активацию эндотелия, способствуя экспрессии eNOS [92, 94]. При этом, miR-125a также снижает поглощение липидов посредством стимулированных макрофагов [92]. Экспрессия miR-223 негативно регулирует синтез холестерина и оказывает противовоспалительное действие за счет ослабления продукции провоспалительных цитокинов [96]. Напротив, miR-92a, миРНК, которая уменьшает эндотелиальное воспаление и размер атеросклеротических бляшек, активируется в бляшках посредством регуляции Kruppel-подобного фактора 2 (KLF2) [96]. Некоторые миРНК также модулируют старение ЭК. Так, miR-let-7g ингибирует клеточное старение, регулируя антивозрастной ген сиртуин 1 (SIRT1) и путь метаболизма инсулинового фактора роста 1 (IGF1) [97], в то время как miR-126a, как было обнаружено, индуцирует старение ЭК посредством сигнальной активации NF-kB [98].
Известно, что микроРНК также регулируют воспалительную реакцию макрофагов при развитии атеросклероза. К ним относятся, в первую очередь, miR-155, miR-222, miR-424, miR-503, miR-146a/b и miR-147. Сверхэкспрессия miR-125b индуцирует активацию макрофагов путем подавления интерферон-регуляторного фактора 4 (IRF4) [96]. Кроме того, было обнаружено, что miR-342-p усиливает провоспалительные макрофагальные факторы, такие как ИЛ-6. На животных моделях показано, что ингибирование miR-342-p уменьшает образование атером, в то время как ингибирование miR-33, регулятора ABCA1 и ABCG1, уменьшает отток холестерина в ЛПВП в макрофагах и способствует развитие атеросклеротических бляшек у мышей [13, 99].
Изменение пролиферации ГМКС также регулируется микроРНК. Сверхэкспрессия miR-21 индуцирует переключение ГМКС в сторону синтетического фенотипа, связанного с пролиферацией и продукцией ECM [100]. Сходным образом ингибирование miR-221 и miR-222 снижает скорость пролиферации ГМКС посредством ингибирования c-Kit, p27 (Kip1) и p57 (Kip2) [13, 96]. Синтетический фенотип ГМКС также индуцируется miR-26a посредством модуляции сигнального пути TGF-β [13, 96]. Напротив, miR-143, miR-145 и miR-1 также играют роль в предотвращении фенотипического переключения ГМКС через Kruppel-подобный фактор 4 (KLF4), KLF5 и/или миокардин. Миокардин также способствует контрактильному фенотипу посредством ингибирования PDGF-β miR-29a и miR-24 [13, 96]. Сходным образом miR-133a способствует сократительному фенотипу в ГМКС, и ее уровни снижаются при атеросклерозе [96]. Кроме того, дифференцировка ГМКС также индуцируется miR-663, которая способствует экспрессии фактора транскрипции JunB и легкой цепи 9 миозина. Также, нацеливание miR-15b и miR-16 на онкогенный yes-ассоциированный белок (YAP) индуцирует более сократительный фенотип в ГМКС. Наконец, несколько других миРНК ассоциированы с контрактильным и синтетическим фенотипом в ГМКС. Таким образом, микроРНК, ассоциированные с контрактильным фенотипом, включают miR195, miR-638, miR-7d, miR-100, miR-10a, miR-204, miR-199a и miR-424; ассоциированные с синтетическим фенотипом миРНК включают miR146a, miR-208 и miR-31. Однако, микроРНК, относящиеся к синтетическому фенотипу, обоснованно относят к антиатерогенным [13, 101].
Механизмы, участвующие в разрыве бляшки, до конца не изучены, но уязвимость бляшки связана с толщиной фиброзной покрышки, развитием некротического ядра и воспалительной реакцией. Однако появляется все больше доказательств того, что миРНК играют роль в процессах, ведущих к разрыву бляшек. МiR-322 активируется в уязвимых бляшках по сравнению со стабильными бляшками, и ее ингибирование приводит к подавлению провоспалительного цитокина ИЛ-6 [101]. МiR-712 также приводит к разрыву бляшки, так как активирует дезинтегрин и металлопротеиназу с тромбоспондиновыми мотивами 4 (ADAMST4) [13, 101]. Как упоминалось ранее, толщина фиброзной покрышки зависит от синтеза и деградации ВКМ и связана с уязвимостью бляшек. ВКМ состоит в основном из коллагенов, которые расщепляются MMP, и было показано, что некоторые миРНК нацелены на MMP (т.е. miR-24 и miR-133a) [93, 102]. МiR-494, которая экспрессируется в атеросклеротических бляшках, подавляет TIMP3 и снижает содержание коллагена в бляшках [96]. Более того, miR-29 помогает поддерживать целостность фиброзной покрышки, нацеливаясь на IFN-γ, который активирует гены проколлагена в ГМКС [92]. Ингибирование генов, которые продуцируют белки ВКМ, с помощью miR-29 может привести к разрыву бляшки [96]. Что касается воспаления в некротическом ядре, было показано, что miR-21 способствует эффероцитозу и подавляет врожденный иммунный ответ. Кроме того, miR-223 как негативный регулятор инфламмасомы NLRP3 предотвращает связанный с ней воспалительный ответ [96]. МiR-155 также способствует формированию некротического ядра при атеросклерозе, что приводит к образованию уязвимых бляшек за счет индукции апоптоза [96, 103]. Кроме того, miR-365 индуцирует эндотелиальный апоптоз, способствуя разрыву бляшек [103]. Несмотря на то, что некоторые микроРНК считаются ключевыми молекулами при атеросклерозе, необходимы их дополнительные исследования, чтобы выяснить точную потенциальную роль миРНК в качестве мишеней для терапии.
Считается, что расшифровка роли различных миРНК как прямых или непрямых посттранскрипционных регуляторов функции ГМКС и ЭК представляет собой серьезную проблему в области патофизиологии гладких мышц и сосудов [13, 103]. Открытие того, что экспрессия miR-143/145 имеет решающее значение для поддержания сократительного фенотипа ГМКС [31, 103], выявило роль бицистриновой единицы, кодирующей miR-143 и miR-145, в регуляции физиологии гладких мышц [103]. Роль miR-143/145 была продемонстрирована на мышах с генетическим дефицитом miR-143/145, у которых снижен сосудистый тонус и контроль артериального давления [13, 31]. Кроме того, функциональная значимость miR-143/145 в патологии сосудов человека была подтверждена наблюдаемым подавлением кластера miR-143/145 в аневризмах аорты человека по сравнению с нормальной тканью аорты [13, 31, 96].
Несколько исследований показали, что клетки могут высвобождать миРНК, которые затем могут проявлять свои специфические эффекты, модулируя процессы в клетках-реципиентах. Так, было показано, что ГМКС связываются с ЭК через miR-143/145 [104]. Межклеточный контакт ГМКС/ЭК индуцирует транскрипцию miR-143/145 в ГМКС, способствуя переносу этих миРНК в эндотелий [104]. В частности, ГМКС могут доставлять miR-143/145 в ЭК через мембранные нанотрубки или туннельные нанотрубки [104]. При этом, перенос miR-143/145 из ГМКС в ЭК стимулируется TGF-β, секретируемым ЭК. Кроме того, miR-143/145, полученная из ГМКС, снижает ангиогенный потенциал ЭК путем репрессии гексокиназы II и интегрина β8 [104]. С другой стороны, miR-126 действует как межклеточный мессенджер, в основном высвобождаемый ЭК и интернализируемый главным образом моноцитами и ГМКС [92, 96]. Не вызывает сомнений, что miR-126 играет критическую роль в модуляции развития сосудов и гомеостаза, нацеливаясь на специфические мРНК, включая белок 1, родственный Sprouty (SPRED-1), CXCL12, SDF-1 и регуляторную субъединицу киназы 2 фосфоинозитол-3 (PIK3R2). Кроме того, miR-126 также связана с эндотелиальной дисфункцией, связанной с развитием диабета и его осложнений [13, 105].
Прогресс, достигнутый технологиями секвенирования нового поколения, выявил увеличение количества днРНК, связанных с патогенезом атеросклероза [13, 106]. Несколько днРНК, идентифицированных в тканях бляшек, играют защитную роль при сосудистых заболеваниях. Так, днРНК MeXis (LXR-индуцированная последовательность, экспрессируемая макрофагами) участвует в транспорте холестерина. Животные модели, лишенные MeXis, показали повышенную атеросклеротическую нагрузку и сниженную экспрессию атерозащитного белка ABCA1 в атеромных бляшках [106]. Снижение экспрессии днРНК MALAT1 (транскрипт 1 метастаз-ассоциированной аденокарциномы легкого) также было связано с развитием атеромных бляшек [107]. Нокдаун MALAT1 способствует поглощению липидов пенистыми клетками, индуцируя транскрипцию рецептора-мусорщика CD36 [13, 106]. Кроме того, lncRNA CHROME (регулятор гомеостаза холестерина экспрессии микроРНК) защищает от атеросклероза, стимулируя отток холестерина, путем ингибирования микроРНК, таких как miR-33 [108]. Однако лишь немногие днРНК проявляют атеропротективные эффекты путем ингибирования апоптоза и старения. В частности, днРНК CERNA1 способствует стабилизации бляшек вследствие ингибирования клеточного апоптоза за счет индукции экспрессии API5, а днРНК SNHG12 в бляшках атером свиней и человека ингибирует повреждение ДНК и старение [108]. В атеросклеротических бляшках сонных артерий человека отмечены пониженные уровни днРНК NEXN-AS1 и днРНК MANTIS [109, 110]. NEXN-AS1 активирует экспрессию NEXN, гена, который оказывает атеропротективное действие в ГМКС и ЭК [109]. Кроме того, одна днРНК, называемая SENCR (гладкомышечная и ЭК-обогащенная миграция/дифференциация-ассоциированная длинная некодирующая РНК), играет роль в развитии атеросклероза, защищая эндотелиальный слой [111]. Так, Boulberda et al. обнаружили измененные уровни днРНК SENCR в сосудистой ткани [112]. Установлено, что недавно идентифицированная днРНК RP11-714G18.1 также подавляется в атеросклеротических бляшках. Эта днРНК ингибирует миграцию ГМКС и ЭК путем подавления экспрессии MMP1, ингибируя прогрессирование атеросклероза. Кроме того, RP11-714G18.1 ингибирует адгезию моноцитов к ЭК [13, 111].
Недавнее исследование подчеркнуло значимую роль днРНК SMILR в атеросклерозе и показало, что ее экспрессия значительно выше в нестабильных бляшках, чем в стабильных бляшках, и увеличивает пролиферацию ГМКС путем регуляции экспрессии проксимального гена HAS2 [113]. Более того, днРНК SMILR напрямую связывается с мРНК митотического белка CENPF (центромерный белок F), управляя пролиферацией гладкомышечных клеток [113]. Аберрантная пролиферация и миграция ГМКС являются критическими факторами в формировании атеросклеротических бляшек, и несколько молекулярных механизмов, которые включают днРНК, контролируют эти процессы. Среди днРНК, подавляющих миграцию ГМКС, днРНК RP11-714G18.1 действует путем прямого нацеливания на ЛПНП-родственный рецептор 2, связывающий белок (LRP2BP) при атеросклерозе [13, 111]. Совсем недавно было обнаружено, что днРНК ZNF800, которая в высокой степени экспрессируется в тканях атеросклеротических бляшек человека и преимущественно в ГМКС, подавляет пролиферацию и миграцию ГМКС через сигнальный путь AKT/mTOR/HIF-1α путем активации PTEN [114]. Другое исследование показало, что экспрессия днРНК RNCR3 значительно выше при атеросклеротических поражениях, что приводит к снижению пролиферации и миграции ГМКС через регуляторную сеть RNCR3/Kruppel-подобного фактора 2/miR-185-5p [115]. Более того, днРНК RP11-714G18.1 нарушает миграцию ГМКС при атеросклерозе посредством LRP2BP-опосредованного подавления MMP1 [13, 111]. Экспрессия днРНК p21 также снижена в атеросклеротических бляшках, и она подавляет пролиферацию ГМКС и атеросклероз за счет усиления активности TP53, тем самым играя атеропротективную роль при развитии атеросклероза [13, 116].
Аналогичным образом днРНК CCL2 активируется в нестабильных атеросклеротических бляшках по сравнению со стабильными атеросклеротическими бляшками. Более того, днРНК CCL2 модифицирует уровни мРНК провоспалительного хемокина CCL2 (или MCP-1) путем взаимодействия с РНК-связывающими белками (HNRNPU и IGF2BP2) [117]. В последнее время днРНК GAS5 (специфическая задержка роста 5) привлекла внимание как потенциальный биомаркер атеросклероза [118]. Уровни GAS5 были повышены в атеросклеротических бляшках, а GAS5 связывает и подавляет микроРНК miR-221, увеличивая продукцию ММР и провоспалительных молекул в атеросклеротических бляшках [119]. Arslan и др. выявили повышенную регуляцию днРНК MIAT (транскрипт, ассоциированный с инфарктом миокарда) в атеросклеротических бляшках [107]. MIAT увеличивает пролиферацию ГМКС путем связывания и подавления miR-181b [120]. Другой днРНК, оказывающей влияние на пролиферацию и миграцию ГМКС, является BANCR (BRAF-регулируемая днРНК 1) [121]. Наконец, днРНК ANRIL (антисмысловая некодирующая РНК в локусе INK4) также является важной молекулой в атерогенезе, так как она влияет на несколько типов клеток в атеросклеротических бляшках, где ее экспрессия активируется и прямо коррелирует с тяжестью атеросклероза [122]. Недавно были выявлены кольцевые формы ANRIL (circANRIL), противоположные линейной форме и включающие разные экзоны. И наоборот, circANRIL обратно коррелирует с риском атеросклероза [123]. CircANRIL был обнаружен в сосудистой ткани, гладкомышечных клетках и макрофагах, где он проявляет атеропротективную функцию [122].
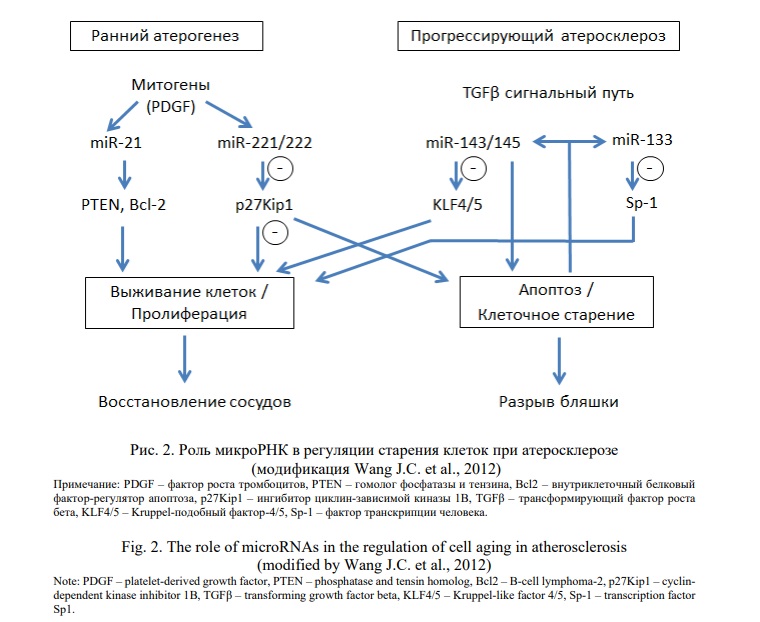
МикроРНК регулируют пролиферацию клеток, старение клеток и апоптоз несколькими путями. Митогены, такие как PDGF, индуцируют активацию miR-21, miR-221 и miR-222, что приводит к выживанию клеток (посредством Bcl2 и гомолога фосфатазы и тензина [PTEN]) и пролиферации клеток посредством ингибирования p27kip1 [124]. В конечном итоге это может способствовать пролиферации ГМКС, например, при раннем атеросклерозе или при восстановлении сосудов. Напротив, ГМКС из старых сосудов в атеросклеротических бляшках демонстрируют сниженный ответ на митогены, сопровождающийся активацией сигнальных путей TGFβ. Увеличение TGFβ на поздних стадиях атеросклероза усиливает экспрессию miR-143/145, которая впоследствии ингибирует Kruppel-подобный фактор-4/5 (KLF4/5) и, следовательно, запрещает пролиферацию ГМКС [124]. Ускоренная остановка клеточного цикла и старение в ГМКС могут стимулировать miR-133, которая дополнительно подавляет пролиферативный фенотип ГМКС и образует петлю обратной связи. Таким образом, происходит ингибирование пролиферации ГМКС, что способствует клеточному старению, признаки, которые, по прогнозам, будут способствовать разрыву бляшки и/или угнетать восстановление бляшек (Рис. 2).
Таким образом, исследования последних лет свидетельствуют о том, что днРНК играет решающую роль в развитии атеросклеротических бляшек, в т.ч. в периоде старения. Полученные знания и продолжающиеся исследования функций некодирующих РНК в развитии бляшек показывают, что микроРНК и длинные некодирующие РНК могут изменять транскрипцию генов, вовлеченных в развитие атеросклероза. Однако для большинства из них специфический механизм действия до конца не ясен, и необходимы дальнейшие исследования, чтобы понять сложную роль регуляторных РНК при атеросклерозе.
9. Возрастзависимые аспекты эволюции атеросклеротических бляшек
Во всем мире вклад сердечно-сосудистых заболеваний в заболеваемость и смертность пожилых людей (в возрасте старше 65 лет) неуклонно растет [16, 125]. Большое значение в этом играют особенности питания и образа жизни, однако с возрастом изменяются также многие физиологические процессы, увеличивающие сердечно-сосудистые риски. В этой связи у пожилых людей менее эффективными могут быть методы лечения таких заболеваний. Понимание механизмов, с помощью которых старение способствует развитию сердечно-сосудистых заболеваний, связанных с атеросклеротическими изменениями, может иметь фундаментальное значение для разработки новых методов лечения возрастных пациентов.
На сегодняшний день принято считать, что старение клеток связано с необратимой потерей их пролиферативной способности. Обычно старение клеток обусловлено истощением репликативного потенциала, например, укорочением теломер, однако, этот процесс может развиваться и как реакция на стресс – так называемое «стрессиндуцированное преждевременное старение» [126]. Оба типа старения характеризуются выходом из клеточного цикла и рядом маркеров, включая активацию ингибитора циклин-зависимой киназы p16ink4a и секрецию ряда цитокинов как часть «секреторного фенотипа, связанного со старением» (SASP) [126, 127]. Так, показано, что атеросклеротические бляшки мышей с нулевым рецептором ЛПНП содержат клетки с активностью рН-чувствительного лизосомального фермента β-галактозидазы (SAβG), связанного со старением, а также мРНК, кодирующей образование цитокинов SASP [126, 127]. SAβG-положительные клетки имели некоторые ультраструктурные особенности эндотелиальных клеток, гладкомышечных клеток сосудов (ГМКС), макрофагов и присутствовали в высоких концентрациях при запущенных поражениях, а также были обнаружены в областях, предрасположенных к атеросклерозу всего 9 дней спустя после жировой диеты. В этих исследованиях использовали трансгенных мышей, содержащих репортерные и/или «суицидальные» гены, необходимые для идентификации и смерти клетки на основании активности промотора p16ink4a [127]. При этом, удаление клеток с активностью промотора p16ink4a уменьшало как образование, так и прогрессирование бляшек, а также способствовало развитию некоторых признаков стабильных бляшек [127].
Ряд исследований наглядно продемонстрировал, что зрелые атеросклеротические бляшки человека имеют признаки старения, включая укорочение теломер и активность SAβG [127], и что индукцию старения в ГМКС человека опосредует IL-1α-зависимый SASP [128]. Однако SAβG не специфичен для стареющих клеток мышей и людей, поскольку он также может быть свойственен макрофагам [129]. Действительно, большинство SAβG-положительных клеток в бляшках человека, по-видимому, представляют собой макрофаги в ядре поражения, с очень редкими интенсивно-положительными ГМКС в фиброзной «шапочке», которые также обнаруживают другие маркеры старения, такие как p16ink4a, но не имеют признаков старения в срединной оболочке [127, 128]. Кроме того, компоненты SASP, используемые исследователями в качестве маркеров старения, такие как матриксные металлопротеиназы, фактор некроза опухоли и IL-1α, все хорошо экспрессируются макрофагами [127]. Промотор p16ink4a экспрессируется в резидентных и воспалительных макрофагах, включая богатые макрофагами области атеросклеротических бляшек человека, а также активируется в момент дифференцировки моноцитов в макрофаги, что происходит при развитии атеросклероза, а также может регулировать поляризацию и способствовать передаче воспалительных сигналов в макрофагах [126, 130]. Существует предположение, что макрофаги определённо способствуют проявлению SAβG и могут быть уничтожены с помощью «суицидных» генов, управляемых промоторами p16ink4a [129]. Вместе с тем, считается, что точную частоту, происхождение и функциональные последствия старения клеток при атеросклерозе еще предстоит определить [126].
Старение в костном мозге искажает дифференцировку гемопоэтических клеток в сторону миелоидных клеток, а также способствует образованию клонов кроветворных клеток без явного развития гемобластозов или других известных клональных нарушений – феномен, известный как клональное кроветворение неопределенного потенциала (CHIP) [131, 132]. Клинические исследования последнего десятилетия показали, что наличие CHIP увеличивает риск развития сердечно-сосудистых заболеваний [131, 133]. Важно отметить, что размер клона CHIP, определяемый как частота варианта аллеля (VAF), коррелирует с риском сердечно-сосудистых заболеваний [133]. В частности, у лиц с клоном CHIP с VAF более 10% риск сердечно-сосудистых заболеваний увеличивается в 12 раз по сравнению с лицами без мутаций, тогда как риск сердечно-сосудистых заболеваний существенно не увеличивается у носителей CHIP с VAF менее 10% [132]. Мутации DNMT3A и TET2 являются самыми распространенными соматическими мутациями, связанными с CHIP [133]. Так, известно, что дефицит TET2, специфичный для миелоидных клеток, увеличивает размер атеросклеротических бляшек [131]. Примечательно, что дефицит TET2 в макрофагах костномозгового происхождения приводит к повышенной секреции IL-6 и IL-1β в ответ на различные стимулы (ЛПНП, ЛПС и IFNγ) [133]. Таким образом, вследствие выявленной связи между IL-6 и CHIP, появилось предположение, что активация воспалительных заболеваний является механизмом, с помощью которого CHIP способствует развитию сердечно-сосудистых заболеваний [133].
По некоторым данным блокада IL-1β снижает риск повторных сердечно-сосудистых событий у пациентов в возрасте старше 60 лет [134]. При этом необходимо отметить, что наибольшая польза от блокады IL-1β наблюдалась у пациентов с низкими уровнями IL-6 в плазме [135]. Результаты этого клинического исследования показывают, что хроническое воспаление является основным фактором возрастного атеросклероза потенциально через передачу сигналов IL-6 [135]. Учитывая этот факт, существует гипотеза, что усиление атеросклероза с возрастом может быть результатом синергии между миелоидными клетками иммунной системы и сосудистой сетью посредством передачи сигналов IL-6 [133].
Отдельный интерес в механизмах развития атеросклероза при старении в ряде исследований представляет повреждение митохондрий [133, 136]. Этот процесс приводит к высвобождению митохондриальных компонентов, известных как молекулярные паттерны, связанные с повреждением митохондрий (mtDAMP), включая митохондриальные ДНК, которые, находясь в цитоплазме, могут активировать внутриклеточные сигнальные пути врожденного иммунитета, такие как ДНК-чувствительный рецептор циклической ГМФ-AМФ-синтазы и инфламмасомы, а также особый воспалительный путь TLR9 [136]. Митохондрии также содержат N-формиловые пептиды, способные повышать хемоаттрактивность нейтрофилов, высвобождать АФК и повреждать артерии [137]. Кроме того, компонент митохондриальных мембран – кардиолипин, тоже может напрямую связываться с NLRP3 и активировать инфламмасому [133]. При хронической активации все эти пути могут способствовать старению сосудов, а также снижать функцию митохондрий, что открывает перспективы дальнейших исследований в этом направлении.
Наконец, существует предположение, что в транссигнальном пути растворимый IL-6R также может вовлекать IL-6 в кровообращение и активировать более широкий спектр клеток, чем в классическом пути, посредством мембранной активации gp130 [133]. Было показано, что ингибирование транссигнализации IL-6 снижает риск развития атеросклероза, подтверждая её возможную патогенную роль в этом процессе [133].
Таким образом, биологическое старение сосудов ускоряется факторами сердечно-сосудистого риска, такими как генетическая предрасположенность. Это приводит к истощению теломер, накоплению повреждений ДНК, увеличению окислительного стресса и эпигенетическим модификациям [124]. Кроме того, наследственные дефекты ферментов репарации ДНК или ламина способствуют ускорению этого процесса. Эпигенетические изменения вызывают транскрипционные изменения, которые могут изменить функцию клеток. Активация реакции на повреждение ДНК (DDR) является одним из наиболее распространенных результатов этих изменений. Сенсоры DDR, такие как ATM и H2AX, фосфорилируются и связываются с поврежденными участками ДНК. Затем добавляются различные белки DDR, такие как MRE11 и NBS1. Эти сигнальные пути запускают ряд нижестоящих эффекторов, таких как p53 и Chk2, что задерживает рост, чтобы восстановить поврежденную ДНК. После успешной репарации клетки могут продолжать пролиферацию и восстанавливать поврежденные сосуды. С другой стороны, неудачная репарация ДНК приводит к накоплению повреждений ядерной и митохондриальной ДНК, зависимому от теломерному и независимому от теломерному старению и апоптозу, а также секретированию провоспалительных цитокинов в рамках секреторного фенотипа, связанного со старением (SASP). Потеря нормальной клеточной функции, а также усиление воспаления при старении и образование атеросклеротических бляшек — все это последствия этих процессов. В результате клеточной дисфункции белки внеклеточного матрикса меняются, что приводит к жесткости и потере эластичности сосудов, а провоспалительные факторы способствуют атеросклерозу. Поскольку восстановление сосудов может способствовать репликативному старению, а провоспалительное состояние и активные формы кислорода (АФК) способствуют преждевременному старению, вызванному стрессом (SIPS), атеросклероз также может напрямую способствовать ускорению старения сосудов (Рис. 3).
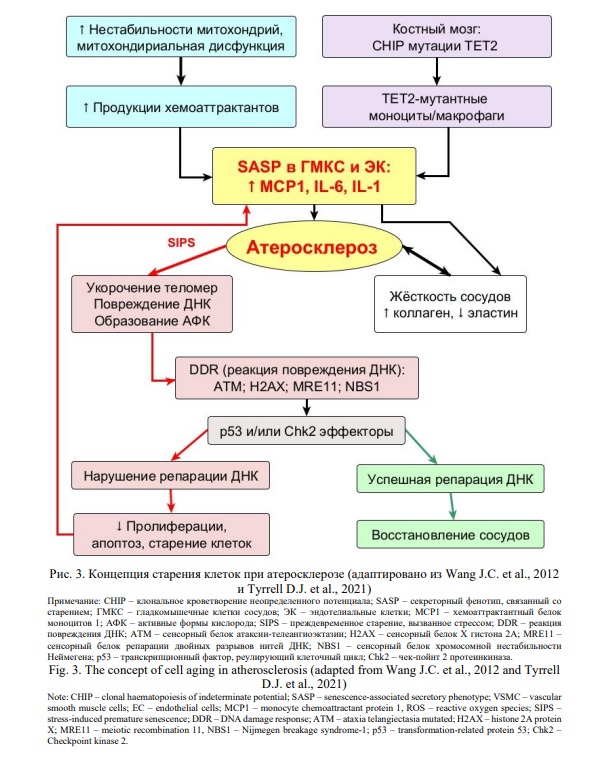
Кроме того, существует программная генетическая теория, которая утверждает, что причиной старения являются точечные мутации в генах, которые определяют продолжительность жизни. Так, генами, потенциально влияющими на скорость старения и связанными с развитием атеросклероза, считают такие гены, как APOE, CHRNA3, WRN, PON1, APOC3, KLOTHO [138]. Но наличие какого-либо аллеля или точечной мутации гена не всегда означает фенотипические проявления вне менделевского наследования. В отсутствие мутаций также существует огромное количество вариантов экспрессии определенных генов. Эпигенетические факторы или «надстройки», регулирующие синтез белка с определенных участков ДНK, являются основной причиной этого. Механизмы Хорвата и Ханнума – одни из самых известных эпигенетических возрастных механизмов, в основе которых лежит метилирование ДНК, биологический процесс, непосредственно связанный со старением. Авторы считают, что в будущем эпигенетические калькуляторы, основанные на часах Ханнума и Хорвата, станут важным инструментом для создания прогностических моделей функционирования клеток, тканей, органов и всего организма [138].
Подводя итог, на данный момент времени можно сделать заключение, что старение влияет на атерогенез несколькими сложными путями, и представляется очевидным, что далеко не один единственный фактор в будущем станет доминирующим патофизиологическим механизмом развития атеросклероза в геронтологии.
Заключение. В течение последних десятилетий наблюдается увеличение встречаемости атеросклеротических поражений сосудов, что способствует риску развития сердечно-сосудистых заболеваний, которые становятся глобальной эпидемией. Изучение клеточных и молекулярно-биологических механизмов атеросклероза позволило лучше понять процессы, приводящие к развитию атеромы, и клинические проявления этого заболевания. Развитие атеросклероза характеризуется накоплением липидов, фиброзных элементов и кальцификацией в крупных артериях, в основе которого лежит активация эндотелия, с последующей вазоконстрикцией и активацией воспалительных механизмов, приводящих в итоге к образованию атеросклеротических бляшек.
Полученные знания и продолжающиеся исследования функций некодирующих РНК в развитии бляшек показывают, что миРНК и днРНК могут изменять транскрипцию генов, вовлеченных в развитие атеросклероза, в т.ч. при клеточном старении. Кроме того, в последнее время появились данные, что микробиота также может быть связана с патогенезом атеросклероза, т.к. выявлены микробные экосистемы в различных средах обитания человека, которые способствуют метаболическим и сердечно-сосудистым нарушениям. Роль микробиоты в развитии атеросклероза подтверждается растущим числом механистических доказательств, однако необходимы дальнейшие исследования, чтобы полнее понять вклад микробиоты в развитие атеросклероза. Наконец, важность анализа половых и возрастных различий как факторов риска, связанных с атеросклерозом, значима для индивидуальных стратегий управления рисками в целях предотвращения развития и прогрессирования атеросклероза. Неуклонный прогресс в понимании механизмов, ведущих к развитию атеросклероза, несомненно, позволит в будущем разработать адекватные методы лечения тяжелых форм течения процесса с постоянным возрастающим риском и, в конечном итоге, улучшит диагностику и прогноз заболевания.
Информация о финансировании
Финансирование данной работы не проводилось.














Список литературы