
Сейпинопатии: клинические варианты наследственной моторно-сенсорной нейропатии и спастической параплегии, обусловленные мутациями в гене BSCL2, у пациентов из Волго-Уральского региона
Aннотация
Актуальность: Нейрональные формы сейпинопатий являются редкими патологиями, и в основном обусловлены двумя патогенными вариантами гена BSCL2 - с.269C>T (p.Ser90Leu) и с.263A>G (p.Asn88Ser). Неполная пенетрантность и субклиническая экспрессивность этих мутаций затрудняют установление генетической этиологии таких случаев болезни. В связи с этим, дальнейшие исследования клинико-генетических особенностей сейпинопатий и их распространенности в различных популяциях являются актуальными. Цель исследования:Установитьгенетическуюпричину заболевания у пациентов с редкими формами наследственных моторно-сенсорных нейропатий (НМСН) и наследственных спастических параплегий (НСП) и охарактеризовать клинический фенотип больных с мутациями в гене BSCL2; определить частоту выявленных мутаций в выборках пациентов с НМСН и НСП из Республики Башкортостан (РБ) – одного из многонациональных регионов Урало-Поволжья. Материалы и методы:В семье с НМСН II поиск генетической причины заболевания проведен методом прямого секвенирования гена BSCL2, в семье с НСП - методом секвенирования экзома таргетной панели генов. Скрининг на наличие/отсутствие выявленных мутаций гена BSCL2 в выборках из других 199 неродственных пациентов с НМСН и 62 пациентов с НСП – жителей РБ, – проведен методом высокочувствительного анализа кривых плавления (HRM). Результаты:У четырех пациентов с НМСН II из одной семьи в генеBSCL2 идентифицирована мутация с.269C>T(p.Ser90Leu) в гетерозиготном состоянии. У всех больных членов этой семьи клиническая картина заболевания соответствовала фенотипу синдрома Сильвера, включающему сочетание признаков пирамидной недостаточности, особенностей нарушения походки и гипотрофии мышц кистей. У пациента с неосложненной формой НСП, а также у его отца с субклиническими проявлениями заболевания обнаружена мутация с.263A>G (p.Asn88Ser). У других неродственных пациентов с НМСН и НСП из РБ данные мутации в гене BSCL2 не обнаружены. Заключение:Полученные данные пополняют сведения о клинико-генетических вариантах сейпинопатий, их геногеографии и распространенности; также они подчеркивают значительную гетерогенность двух групп нейродегенеративных заболеваний – НМСН и НСП, выявляя в их отдельных формах общие механизмы патогенеза и некоторые клинические симптомы, которые могут облегчить точную постановку диагноза
Ключевые слова: сейпинопатии, ген BSCL2, наследственные моторно-сенсорные нейропатии, наследственные спастические параплегии
Введение. Ген BSCL2 (Berardinelli-Seip Congenital Lipodystrophy 2), локализованный в хромосомной области 11q12.3, кодирует сейпин – трансмембранный белок эндоплазматического ретикулума (ЭПР), играющий основную роль в дифференцировке адипоцитов и адипогенезе. В гене BSCL2 на сегодняшний день зарегистрировано 58 различных патогенных вариантов (Human Gene Mutation Database, 2023.4), в большинстве случаев обусловливающих заболевание липидного обмена – врожденную генерализованную липодистрофию Берардинелли-Сейпа типа 2 (CGL2, ОМIM №269700) и реже – патологии нейродегенеративного генеза, к которым относят прогрессирующую энцефалопатию с липодистрофией или без нее (PELD, ОМIM №615924), а также заболевания двигательных нейронов - дистальную моторную нейропатию (dHMN-V, ОМIM №619112), иногда называемую дистальной спинальной мышечной атрофией (dSMA5C, ОМIM №606158), аксональную форму наследственной моторно-сенсорной нейропатии (НМСН II) и одну из форм наследственных спастических параплегий (НСП) – синдром Сильвера (SPG17, ОМIM №270685) [1, 2]. Все заболевания, обусловленные мутациями в гене BSCL2, объединяют под общим названием «сейпинопатии» [3], хотя ранее было выдвинуто предложение называть сейпинопатиями только заболевания двигательных нейронов, связанных с BSCL2 [4].
Наследственные нейропатии (болезнь Шарко-Мари-Тута), в том числе моторно-сенсорные нейропатии (НМСН), и наследственные спастические параплегии (НСП) – это две клинически и генетически гетерогенные группы нервных болезней, для каждой из которых идентифицировано более 100 различных генов, мутации в которых являются причинами соответствующих заболеваний (http://www.neuromuscular.wustl.edu). В обеих группах встречаются генетические формы с различными типами наследования – аутосомно-доминантным, аутосомно-рецессивным, Х-сцепленным, а также с митохондриальным наследованием. Клинические проявления наследственных моторно-сенсорных нейропатий связаны преимущественно с поражением периферической нервной системы, а спастических параплегий – с поражением центрального мотонейрона, при этом, клинические особенности этих патологий и тип наследования могут зависеть не только от самого причинного гена, но и от характера мутаций: различные патогенные варианты в одном и том же гене могут приводить к различным клиническим формам. Известны генетические «перекрывания» НМСН и НСП, связанные с различными мутациями в одних и тех же генах, примером чего в том числе являются и сейпинопатии – клинические варианты этих заболеваний, обусловленные мутациями в гене BSCL2 [5]. Точная дифференциация генетических вариантов, как при НМСН, так и при НСП возможна только на основе ДНК-анализа. Для поиска генетического дефекта наиболее эффективными являются методы NGS – секвенирования (Next Generation Sequensing), но, в то же время, учитывая их достаточно высокую стоимость, часто более рациональным подходом является использование более простых классических методов. В клинической практике в каждом отдельном случае для выбора адекватного метода ДНК-диагностики важными являются наиболее полные сведения о клинико-генеалогическом фенотипе пациента. Знание же молекулярной причины расстройства крайне важно для пациентов и членов их семей, поскольку устраняет необходимость в постоянном поиске возможных триггеров болезни и значительно повышает эффективность медико-генетического консультирования: позволяет прогнозировать характер течения заболевания, проводить раннюю – пресимптоматическую и пренатальную, – диагностику.
Заболевания двигательных нейронов, связанные с мутациями в гене BSCL2, были выявлены в разных странах Европы и Азии, но, в целом, их частота невелика и, вероятно, может варьировать в различных регионах мира. К наиболее частым причинам таких сейпинопатий относятся патогенные варианты в двух позициях третьего экзона гена BSCL2 – с.263A>G (p.Asn88Ser) и с.269С>T (p.Ser90Leu) (NM_032667.6). Клиническая картина у пациентов с этими мутациями достаточно гетерогенна, даже в семьях с одинаковой мутацией, поэтому дальнейшее накопление информации о таких случаях, их клинических особенностях и распространенности в различных популяциях представляет определенный научно-практический интерес.
Цель исследования. Установить генетическую причину заболевания у пациентов с редкими формами наследственных моторно-сенсорных нейропатий (НМСН) и наследственных спастических параплегий (НСП) и охарактеризовать клинический фенотип больных с мутациями в гене BSCL2; определить частоту выявленных мутаций в выборках пациентов с НМСН и НСП из Республики Башкортостан (РБ) – одного из многонациональных регионов Урало-Поволжья.
Материалы и методы исследования. Данное исследование было проведено в рамках общего проекта по эпидемиологическому и молекулярно-генетическому изучению наследственных моторно-сенсорных нейропатий и спастических параплегий в Республике Башкортостан. Банк ДНК сформирован на основе данных о больных, состоящих на диспансерном учете в Республиканском медико-генетическом центре (РМГЦ). Больные и их ближайшие родственники наблюдаются специалистами – нейрогенетиками, а также сотрудниками кафедры неврологии Башкирского государственного медицинского университета. ДНК выделена методом фенольно-хлороформной экстракции из цельной венозной крови.
На сегодняшний день наша коллекция ДНК «НМСН» включает образцы 312 больных и 100 их близких родственников без клинических симптомов заболевания из 200 неродственных семей (71 – русских, 56 – татарских, 38 – башкирских, 23 – метисных и других национальностей). Во всех 200 семьях, в соответствие с типом НМСН, ранее был проведен поиск мутаций в генах PMP22, MPZ, GJB1, EGR2, MFN2, GDAP1, NEFL, наиболее часто связанных с заболеванием. Мутации в этих генах были идентифицированы в 121 семье (60,5%). Коллекция ДНК «НСП» включает образцы 76 пациентов и 40 их близких родственников без клинических симптомов заболевания из 63 неродственных семей (27 –татарских, 14 –русских, 5 – башкирских, по одной семье – чувашской, украинской; марийской, 8 – метисных семей, а также 6 семей с неустановленной этнической принадлежностью). В этих 63-х семьях с НСП ранее были исследованы гены SPAST, ATL1A, REEP1 и NIPA1, мутации в которых были идентифицированы в 28 семьях (44%).
Патогенный нуклеотидный вариант с.269C>T в гене BSCL2 был идентифицирован у пациентки с НМСНII в результате целенаправленного исследования этого гена методом прямого секвенирования по Сэнгеру. Патогенный вариант с.263A>G был выявлен методом массового параллельного экзомного секвенирования у пациента с НСП с отсутствием мутаций в указанных выше генах. Это исследование было проведено на секвенаторе нового поколения Ion S5. Для пробоподготовки использована технология ультрамультиплексной ПЦР, сопряженная с последующим секвенированием (AmpliSeq™). Анализ проведен с использованием кастомной панели «Спастические параплегии», включающей кодирующие последовательности генов: GJC2, AP4B1, AMPD2, IBA57, ALDH18A1, ZFYVE27, NT5C2, ENTPD1, MTPAP, CAPN1, BSCL2, KLC2, KIF5A, C12orf65, MARS, VAMP1, B4GALNT1, SPG20, SACS, ATL1, ZFYVE26, DDHD1, TECPR2, AP4S1, NIPA1, SPG11, SPG21, AP4E1, USP8, SPG7, FA2H, ARL6IP1, KIF1C, AFG3L2, RTN2, PNPLA6, C19orf12, CPT1C, MAG, HSPD1, KIF1A, REEP1, PGAP1, MARS2, SPAST, SLC33A1, TFG, WDR48, CYP2U1, ARSI, ZFR, REEP2, AP5Z1, AP4M1, CYP7B1, KIAA0196, ERLIN2, VPS37A, DDHD2, GBA2, L1CAM, PLP1, SLC16A2. По данным AmpliSeq™ Сoverage Analysis, среднее покрытие панели «Спастические параплегии» составляет 363.1, Uniformity 92,02%. Обработка данных секвенирования проведена с использованием стандартного автоматизированного алгоритма, предлагаемого TermoFisher Scientific (Torrent Suite™), а также программного обеспечения Gene-Talk. Для оценки популяционных частот выявленных вариантов использованы выборки проектов «1000 геномов», ESP6500 и Genome Aggregation Database (gnomAD). Для оценки клинической релевантности выявленных вариантов использованы база данных OMIM (https://www.ncbi.nlm.nih.gov/omim), база данных по патогенным вариантам HGMD® Professional версия 2023.4 (https://www.hgmd.cf.ac.uk/ac/uk), специализированные базы данных по соответствующим заболеваниям (https://neuromuscular.wustl.edu/time/hmsn.html) и литературные данные.
Патогенные варианты, выявленные у пробандов, были идентифицированы также у их близких родственников методом секвенированием по Сэнгеру: для амплификации участка 3-го экзона гена BSCL2 праймеры подобраны с помощью программы Primer3 (v.0.4.0) (http://fokker.wi.mit.edu/primer3/input.htm); секвенирование проведено на автоматическом анализаторе ABI PRISM 3500 XL (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA). Для дальнейшего поиска нуклеотидных замен с.269C>T и с.263A>G в гене BSCL2 в группах других больных из обследованных выборок НМСН и НСП был использован метод высокочувствительного анализа кривых плавления (HRM); анализ проведен на термоциклере для амплификации нуклеиновых кислот LightCycler 96 (Roche, Швейцария). Скрининг на наличие/отсутствие данных мутаций был проведен у всех неродственных пациентов в каждой из выборок (200 – с НМСН, 63 – с НСП), в том числе у пациентов с выявленными мутациями в других генах. Такой подход был использован для того, чтобы повысить точность оценки частоты исследуемых мутаций в группах больных из обследуемого региона, поскольку известны случаи выявления у пациентов одновременно двух мутаций в различных генах, приводящих к развитию одного и того же заболевания.
Все процедуры, выполненные в исследовании с участием людей, соответствуют этическим стандартам институционального и/или национального комитета по исследовательской этике и Хельсинкской декларации 1964 г. и ее последующим изменениям или сопоставимым нормам этики. От каждого из включенных в исследование участников было получено информированное добровольное согласие. Исследования были одобрены биоэтическим комитетом ИБГ УФИЦ РАН.
Результаты исследования. У пациентки-пробанда с аутосомно-доминантной моторно-сенсорной нейропатией II типа (семья НМСН – М.263, русской этнической принадлежности) методом прямого секвенирования кодирующих последовательностей гена BSCL2 был выявлен ранее описанный патогенный вариант с.269C>T (p.Ser90Leu) в гетерозиготном состоянии (Рис. 1), впоследствии обнаруженный также у троих ее детей – у дочери и у двух сыновей – монозиготных близнецов. Другие родственники, к сожалению, не были доступны для данного анализа.
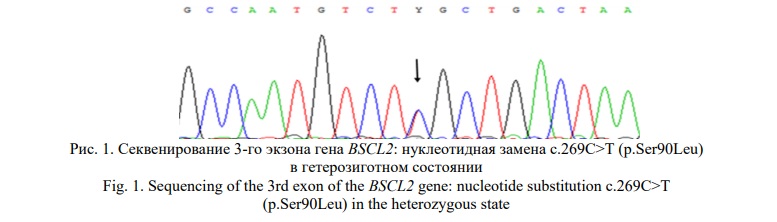
Семья М.263 наблюдается в медико-генетической консультации с диагнозом: наследственная моторно-сенсорная нейропатия II типа. Изменение походки – «ходьбу на носочках» родители пробанда Н.М. начали отмечать с 4-летнего возраста дочери и за последующие годы, несмотря на регулярное лечение, назначенное неврологом, походка у девочки продолжала ухудшаться. Генетиком при первом осмотре пробанда в возрасте 12 лет был предположен вариант наследственной спастической параплегии, так как были отмечены нарушение походки с опорой на передние отделы стоп, затруднение ходьбы на пятках и приседания на полную ступню, легкое повышение тонуса по спастическому типу в мышцах – разгибателях голеней, повышение сухожильных рефлексов в руках и ногах, положительные стопные знаки Россолимо и Бабинского с обеих сторон. Мать пробанда оказалась фенотипически здоровой, сообщила о измененной походке у отца девочки и его матери – бабушки по отцовской линии. Клинический фенотип пробанда в возрасте 28 лет соответствовал проявлениям моторной полинейропатии: изменение походки по типу «степпажа», деформации стоп по типу «полых», ретракция ахилловых сухожилий с невозможностью приседания на полную ступню, мышечные гипотрофии в кистях рук, стопах, голенях со снижением мышечной силы преимущественно в мышцах - разгибателях: стоп – до 2 баллов, кистей – до 4 баллов. Сухожильные рефлексы рук и коленные рефлексы были повышены, а ахилловы – отсутствовали, чувствительных расстройств выявлено не было. С учетом электрофизиологических показателей (значительное снижение амплитуды М-ответов (до 1 мВ) с двигательных волокон нервов рук и ног при скорости распространения возбуждения от 45 до 68 м/с) был установлен клинический диагноз аксональной наследственной моторно-сенсорной нейропатии (НМСН II типа). В последующие осмотры, проведенные в возрасте 31 и 40 лет, у пробанда отмечены медленное нарастание гипотрофий (особенно мышц кистей) и слабости в мышцах ног и рук, сохраняющаяся гиперрефлексия коленных рефлексов и сухожильных рефлексов с рук, прогрессирующая эквиноварусная деформация стоп (в возрасте 31 года проведена экзартикуляция 1 пальца правой стопы вследствие остеомиелита, развившегося в результате нагноения натоптыша), снижение поверхностной чувствительности в стопах. При повторном электронейромиографическом исследовании (возраст пробанда 40 лет) М-ответы с двигательных волокон нервов ног не регистрировались, с нервов рук были снижены до 0,05-0,1 мВ, по двигательным волокнам нервов рук и ног скорость распространения возбуждения составила от 28 до 40 м/с; по сенсорным волокнам нервов ног – 41-56 м/с при нормальных показателях амплитуды.
Дочь пробанда, А.М., была впервые осмотрена в возрасте 6 лет; из анамнеза известно, что изменение походки («начала косолапить») и частые спотыкания появились в возрасте 4 лет. При осмотре выявлены затруднение ходьбы на пятках, снижение ахилловых рефлексов при одновременном повышении коленных рефлексов и сухожильных рефлексов с рук; патологических стопных знаков и нарушений чувствительности не было. В 8-летнем возрасте добавилось изменение походки по типу «степпаж». Следующие осмотры были проведены в возрасте 20 и 22 лет, к жалобам добавилась слабость и похудание мышц ног и рук, а также прогрессирующее ухудшение ходьбы. В клинической картине отмечались дистальные мышечные гипотрофии, снижение мышечной силы, преимущественно в мышцах - разгибателях стоп (до 2 баллов) и кистей (до 3 баллов), гиперрефлексия с рук, повышение коленных рефлексов, отсутствие ахилловых рефлексов, положительные патологические знаки Бабинского и Россолимо с обеих сторон, легкое снижение поверхностной чувствительности в стопах. При электронейромиографическом исследовании (возраст пробанда 21 год) выявлено снижение амплитуды М-ответов по двигательным волокнам нервов ног – 0,5-1,0 мВ, нервов рук – 1,5-4,5 мВ, скорость распространения возбуждения составила 34-39 м/с по нервам ног, 35-72 м/с – по нервам рук. По сенсорным волокнам нервов рук и ног регистрировались нормальные амплитуды ответов и скорости распространения возбуждения, за исключением небольшого снижения скорости по обоим суральным нервам.
Сыновья пробанда – монозиготные близнецы С.М. и А.М. – были осмотрены в возрасте 10 лет. У обоих с 5-летнего возраста регистрировалось нарушение походки, позднее появились жалобы и на слабость в мышцах ног. С.М., в отличие от брата, ходит с опорой на передние отделы стоп («на носочках»), не может стоять на пятках, у него отмечается выраженная ретракция ахилловых сухожилий, частичные контрактуры голеностопных суставов. У А.М. затруднена ходьба на пятках. У обоих братьев есть гипотрофии мышц кистей и снижение их силы до 4 баллов; снижение силы мышц-разгибателей стоп до 3 баллов у С.М. и до 4 баллов у А.М.; у обоих братьев сухожильные рефлексы рук и коленные рефлексы повышены, а ахилловы рефлексы отсутствуют, регистрируются положительные стопные знаки; чувствительных нарушений нет.
В целом, в семье М.263 по результатам электрофизиологического исследования был установлен II тип наследственной моторно-сенсорной нейропатии; в дальнейшем, учитывая особенности фенотипа: признаки пирамидной недостаточности, особенности нарушения походки и отчетливые гипотрофии мышц кистей (особенно мышц тенара и мышц 1 межкостного промежутка), был предположен диагноз сейпинопатии, и в результате анализа гена BSCL2 была выявлена ранее описанная патогенная мутация c.269C>T (p.Ser90Leu).
У другого пробанда – пациента с аутосомно-доминантной спастической параплегией (семья НСП – Е.47, русской этнической принадлежности), – методом таргетного секвенирования экзома в гене BSCL2 был выявлен в гетерозиготном состоянии другой известный патогенный вариант – с.263A>G (p.Asn88Ser). Методом секвенирования по Сэнгеру (Рис. 2) данный нуклеотидный вариант был подтвержден у пробанда, выявлен у его отца, также имеющего признаки НСП, и исключен у здорового сибса.

Клиническая картина обследованного нами пациента с мутацией с.263A>G (p.Asn88Ser) в гене BSCL2 соответствовала неосложненной форме НСП. Впервые этот пациент в возрасте 17 лет обратился с жалобами на скованность в мышцах нижних конечностей, необходимость расхаживаться, что беспокоило его в течение последних трех лет. В неврологическом статусе: нижний спастический парапарез с повышенным мышечным тонусом до 1-2 баллов по шкале Ашворт.
Отец пробанда при осмотре не имел жалоб, но в неврологическом статусе были субклинические проявления заболевания в виде оживления глубоких сухожильных рефлексов, наличия патологических стопных знаков.
Для поиска мутаций с.269С>Т и с.263A>G в гене BSCL2 могут быть использованы различные методы. Так, методом ПДРФ-анализа мутация с.269С>Т может быть идентифицирована с помощью эндонуклеазы BsmAI, для которой при наличии нуклеотидной замены существующий в норме сайт рестрикции исчезает. Мутация с.263A>G идентифицируется с помощью эндонуклеаз BsrI и TspRI, для которых при наличии нуклеотидной замены появляется сайт рестрикции, отсутствующий в норме. В нашем исследовании для скрининга данных мутаций в группах неродственных больных НМСН и НСП мы использовали HRM – анализ, как более удобный метод, не требующий наличия рестриктаз, позволяющий идентифицировать оба интересующие нас варианта (Рис. 3).
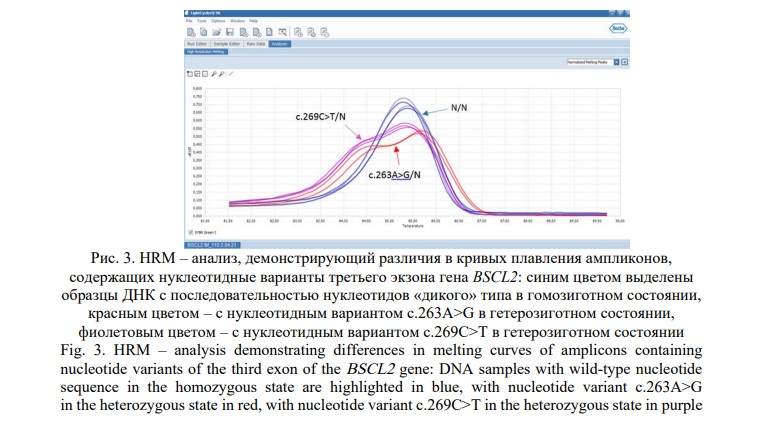
В результате проведенного скрининга у других пациентов с НМСН и НСП, не родственных описанным выше пробандам, не было выявлено изменений кривых плавления, отличных от нормы, и, соответственно, искомые нуклеотидные замены в третьем экзоне гена BSCL2 обнаружены не были. Таким образом, частота мутации с.269C>T (p.Ser90Leu) в общей выборке неродственных пациентов с НМСН из РБ составила 0,5% (1/200), а среди русских пациентов – 1,4%; частота мутации с.263A>G (p.Asn88Ser) в общей выборке неродственных пациентов с НСП составила 1,6% (1/63), а среди русских больных – 7%.
Обсуждение. Кодируемый геном BSCL2 белок сейпин участвует в биогенезе липидных капель (ЛД) – органелл для хранения триацилглицеролов в жировой ткани, играющих ключевую роль в гомеостазе липидов и нормальной физиологии клеток [6]. В частности, было установлено, что сейпин образует мультимерные фокусы и, являясь частью белкового механизма, действующего в местах контакта ЭПР-ЛД, способствует росту зарождающихся ЛД. При дефиците сейпина образующиеся в клетках липидные капли имеют неправильный, неоднородный, размер и аномальный состав липидов и белков, что, вероятно, приводит к клеточной дисфункции в отношении хранения и восстановления липидов для клеточных нужд [7]. Ген BSCL2 широко экспрессируется в различных тканях человека, но самые высокие уровни его экспрессии обнаруживаются в жировой ткани, головном мозге и яичках [8, 9]. В центральной нервной системе этот ген экспрессируется не только в головном, но и в спинном мозге [10, 11]. Детальное обсуждение физиологических функций сейпина, механизмов его участия в процессе адипогенеза и в развитии заболеваний человека представлено в недавних обзорах [12, 13].
Аномальная экспрессия сейпина, обусловленная мутациями в гене BSCL2, может приводить к повреждению различных органов и развитию ряда наследственных заболеваний. Дефицит сейпина приводит к развитию врожденной генерализованной липодистрофии 2 типа, характеризующейся почти полной потерей жировой ткани и тяжелыми метаболическими нарушениями [8, 14]. С накоплением токсичных олигомеров сейпина связывают развитие нейрональных нарушений [15]. Патологиями нейродегенеративного генеза являются прогрессирующая энцефалопатия с липодистрофией или без нее – фатальное детское заболевание, характеризующееся регрессом развития двигательных и когнитивных навыков в первые годы жизни [3, 16], и BSCL2-ссоциированные заболевания двигательных нейронов.
Различия в структурно-функциональных нарушениях белка сейпина связаны с различным характером мутаций в гене BSCL2. Структурно, в белке сейпина его N- и С- концы экспонированы в цитоплазму, а две трансмембранные спирали в просвете ЭПР соединяются с большой, эволюционно консервативной люминальной петлей, содержащей мотив N- гликозилирования (аминокислотные остатки 31-252) [17, 18]. Мутации, нарушающие процесс гликозилирования сейпина, в том числе с.263A>G (p.Asn88Ser) и c.269C>T(p.Ser90Leu), способствуют неполноценной деградации белка убиквитин-протеасомной системой и нарушенной укладке. Накопленные в результате мисфолдинга белковые молекулы провоцируют стресс ЭПР, приводящий к апоптозу нейронов [19, 20, 21].
В настоящее время описано уже достаточно большое число пациентов с мутациями c.269C>T(p.Ser90Leu) и с.263A>G (p.Asn88Ser) в гене BSCL2, но, несмотря на это, установить четкие гено-фенотипические корреляции оказалось достаточно сложно. Было показано, что даже при одинаковых вариантах (p.Asn88Ser или p.Ser90Leu) у разных больных клиническая картина может сильно варьировать как по спектру симптомов, так и по степени тяжести заболевания – от совсем незаметных проявлений у их носителей до выраженных нарушений, приводящих к инвалидности [19, 22-25]. Это подтверждает неполную пенетрантность и субклиническую экспрессию данных патогенных вариантов, а синдром Сильвера и некоторые формы дистальных наследственных моторных нейропатий, в частности, dHMN-V, могут являться крайними фенотипами, имеющими одинаковую генетическую этиологию. Их общей особенностью является выраженная слабость и истощение мышц кисти, которые проявляются на ранних стадиях заболевания и могут преобладать в дальнейшем. Наряду с этим, у пациентов с синдромом Сильвера выявляются спастичность нижних конечностей от легкой до тяжелой, что также указывает на поражение верхних двигательных нейронов. Поражение пирамидного тракта у пациентов с синдромом Сильвера подтверждается и МРТ-исследованиями, демонстрирующими аномалии кортико-спинального тракта на изображениях FLAIR, аналогично другим заболеваниям, связанным с верхним двигательным нейроном, а именно боковому амиотрофическому склерозу [23]. Сенсорные нарушения у пациентов с мутациями p.Ser90Leu и p.Asn88Ser также являются достаточно частыми симптомами заболеваниями, описанными в ряде исследований [25, 26]. В дополнение ко всему сказанному следует также отметить, что синдром Сильвера и dHMN-V сами по себе являются генетически гетерогенными, и некоторые индивидуумы с каждым из этих фенотипов не несут мутацию в гене BSCL2 [19].
В нашем исследовании наиболее детальное описание клинической картины заболевания удалось получить в семье с мутацией p.Ser90Leu. У четырех больных членов этой семьи - пациентки – пробанда, ее дочери и двух сыновей – монозиготных близнецов, - целенаправленные наблюдения по поводу болезни с диагнозом НМСНII проводились на протяжении многих лет их жизни. У них у всех первые признаки заболевания – слабость в ногах и нарушение походки с опорой на передние отделы стоп, - были отмечены в возрасте 4-6 лет. В последующем наблюдалось плавное неуклонное прогрессирование болезни, с развитием клинических признаков, в значительной степени соответствующих таковым при синдроме Сильвера. В частности, у всех выявлялись повышение сухожильных рефлексов в руках и ногах, за исключением ахилловых рефлексов, и положительные стопные знаки; гипотрофия мышц кисти со снижением их силы также была выявлена у всех этих пациентов, но в разном возрасте (зафиксированы в 28, 22 и в 10 лет), хотя точный возраст явного проявления этого признака определить сложно, поскольку обследование не было ежегодным. Спастичность нижних конечностей была отмечена только у пациентки - пробанда, на основании чего первоначально ей был поставлен диагноз «спастическая параплегия». Чувствительные расстройства в виде гипостезии поверхностной чувствительности в стопах были выявлены у пробанда и ее дочери в возрасте после 20 лет, тогда как у сыновей при осмотре их в 10-летнем возрасте таких проявлений отмечено не было. На основании электронейромиографического исследования у всех диагностировалась преимущественно моторная аксональная полинейропатия. В клинической картине монозиготных близнецов выявлены некоторые различия в степени выраженности симптомов, что, вероятно, может быть обусловлено эпигенетическими факторами. Дальнейшее наблюдение за ними и углубленный молекулярный анализ представляют несомненный интерес.
В другой семье, с мутацией с.263A>G (p.Asn88Ser), неврологическое обследование пробанда было проведено единожды, в возрасте 17 лет и, к сожалению, расширенное описание его клинической картины, данные ЭНМГ-исследования отсутствуют. У этого пациента на момент осмотра не было выявлено явных признаков сейпинопатии, а были зарегистрированы только симптомы неосложненной формы НСП. У отца пробанда были выявлены субклинические проявления заболевания. Такая картина, как показывает анализ опубликованных данных, также может наблюдаться при синдроме Сильвера, обусловленном мутацией p.Asn88Ser, но точный генетический диагноз в такой ситуации предсказать сложно.
По распространенности BSCL2-ассоциированных заболеваний двигательных нейронов детальных сведений пока нет. Синдром Сильвера, обусловленный мутацией p.Asn88Ser, ранее был идентифицирован в целом ряде семей из Европы (Англия, Австрия, Германия, Голландия, Италия, Франция, Португалия [19, 22, 23, 24, 27, 28, 29] и Азии (Япония) [25, 30], но общая распространенность этого заболевания считается относительно низкой (<1/100000), и нуклеотидный вариант с.263A>G в гене BSCL2 также регистрируется c низкой частотой – 0,00001591 (Genome Aggregation Database (gnomAD, 2024)). В работах, посвященных данной мутации, было показано ее отсутствие у здоровых лиц из контрольных популяционных выборок (250 австрийцев, 100 бельгийцев, 200 англичан [19], 100 итальянцев [23]). Сравнение гаплотипов в австрийской, итальянской и двух английских семьях с синдромом Сильвера, проведенное Windpassinger et al. (2004), показало, что эти семьи не имеют общих предков, и что в каждой семье мутация p.Asn88Ser возникла независимо. Нуклеотидный вариант c.269C>T(p.Ser90Leu) также был зарегистрирован в различных регионах мира – в семьях пациентов из Бразилии, Бельгии [19, 24], Италии [31], Кореи [32], Китая [33], Тайваня [21], Японии [26], но его общая частота также является низкой, составляет 0,000006573 (gnomAD, 2024). Описаны случаи выявления мутации p.Ser90Leu denovo в семьях с синдромом Сильвера и дистальной наследственной моторной нейропатией [32, 34]. Кроме этих двух относительно частых мутаций, в третьем экзоне гена BSCL2 был зарегистрирован еще один патогенный вариант – с.287G>A (Arg96His), выявленный в Тайване у пациента со спорадической формой дистальной моторной нейропатии [21]. Все эти факты – близкое расположение описанных нуклеотидных вариантов, их широкое географическое распространение и независимое происхождение в разных семьях, случаи denovo, – свидетельствуют об определенной нестабильности соответствующей области третьего экзона гена BSCL2. Учитывая также неполную пенетрантность, можно предположить высокую вероятность повсеместного выявления пациентов с заболеваниями, обусловленными данными мутациями. Описанные в нашем исследовании случаи сейпинопатии среди пациентов с НМСНII и НСП из Республики Башкортостан подтверждают это предположение.
Заключение. Обобщая приведенные данные, можно сделать заключение, что четкие гено-фенотипические корреляции для сейпинопатий пока установить достаточно сложно, как ввиду неполной пенетрантности мутаций, так и вероятного наличия других генетических и эпигенетических факторов, модифицирующих клинический фенотип. Важно также отметить, что несмотря на доминантный характер описанных мутаций в гене BSCL2, из-за их неполной пенетрантности или иногда – субклинической экспрессии, или медленного прогрессирования заболевания, – семейная история сейпинопатий может прослеживаться не всегда. Но и в таких случаях генетический анализ имеет важное значение. Сочетание у пациента определенных клинических проявлений – признаков пирамидной недостаточности, особенностей нарушения походки и гипотрофии мышц кистей, – позволяет заподозрить вариант сейпинопатии и провести целенаправленное исследование гена BSCL2, с первоначальным поиском мутаций в его третьем экзоне. Таким опытом явилось проведенное нами исследование в семье с НМСНII с мутацией p.Ser90Leu, у больных членов которой прослеживались клинические признаки, описанные ранее у пациентов с сейпинопатиями. У пациентов другой семьи, с мутацией p.Asn88Ser, нами были выявлены только симптомы неосложненной формы НСП, не являющиеся характерными для какой-то определенной генетической формы заболевания. Очевидно, что в таких случаях для поиска генетической причины болезни наиболее эффективным методом является NGS-секвенирование. В Республике Башкортостан исследованные патогенные варианты гена BSCL2 являются редкими причинами заболеваний, обнаруженными пока в единичных семьях пациентов с НМСНII и НСП, но, учитывая доминантный характер мутаций, следует ожидать появления в регионе и других аналогичных случаев, и при проведении медико-генетического консультирования обращать внимание также и на происхождение пациентов – их этнос, место жительства, семейную историю в отношении болезни.
Полученные данные дополняют сведения о клинико-генетических вариантах сейпинопатий, их геногеографии и распространенности. Также они еще раз подчеркивают значительную гетерогенность двух групп нейродегенеративных заболеваний – НМСН и НСП, выявляя в их отдельных формах общие механизмы патогенеза и некоторые клинические симптомы, которые могут облегчить точную постановку диагноза.
Информация о финансировании
Работа выполнена в рамках государственного задания Минобрнауки РФ (№122041400169-2), при частичной финансовой поддержке гранта РФФИ (№ 17-44-020951) и Санкт-Петербургского государственного университета (ID PURE: 103964756).
Благодарности
Для исследования использовано оборудование регионального центра коллективного пользования «Агидель» УФИЦ РАН. Образцы ДНК для исследования взяты из «Коллекции биологических материалов человека» ИБГ УФИЦ РАН, поддержанной Программой биоресурсных коллекций ФАНО России (соглашение № 007-030164/2).














Список литературы