
Evaluation of efficacy of selective phenolic compounds of olive leaves in chronic myeloid leukemia: an in-silico approach
Abstract
Background: Chronic myeloid leukemia (CML) is a blood cancer driven by the BCR-ABL1 fusion protein, where current therapies face challenges like resistance and side effects. Olive leaves contain phenolic compounds, which have shown anti-cancer potential. The aim of the study:To use molecular docking analysis to compare the molecular interactions of selected phenolic compounds in olive leaf extract and Imatinib with the active sites of the Breakpoint Cluster Region (BCR)-Abelson (ABL) fusion protein to identify safer and cost-effective therapeutic candidates for Chronic Myeloid Leukemia (CML). Materials and methods: In this work, we assessed the biological activity of the test substances in silico using the PASS online server. ADME analysis was performed utilizing the SwissADME free online web server, and a toxicology study was done using the AdmetSAR online server. For the in silico molecular docking investigation, the protein structure of the BCR-ABL fusion protein was obtained from the Protein Data Bank (PDB) website, while the ligand structures were obtained from the PubChem website. The binding energy (kcal/mol) was determined using the Autodock Vina software. The protein-ligand interactions were examined using the Discovery Studio Visualizer. Results: In silico molecular docking experiments show that verbascoside, uvaol, loganic acid, secologanin, and cinnamic acid have a high binding affinity to the BCR-ABL protein, with binding energies of -9.2, -9.7, -8.7, -7.3, and -7.4, respectively, which are very close to the binding affinity of the standard medication Imatinib, which has a binding energy of -10.3 Kcal/mol. ADMET analysis and PASS prediction validate the following compounds drug-like properties, i.e., anti-carcinogenic, anti-neoplastic, and anti-neoplastic, along with maintaining Lipinski’s rule of five. Conclusion: The binding affinity of some test compounds compared to Imatinib, as well as their interactions with amino acid residues at the active sites of the BCR-ABL fusion protein, suggest that verbascoside, uvaol, loganic acid, secologanin, and cinnamic acid may bind selectively to CML cells, inhibiting their proliferation and acting as a novel anti-CML agent
Keywords: olive leaves, phenolic compound, chronic myeloid leukemia, PASS prediction, SwissADME, molecular docking, Autodock Vina, verbascoside, uvaol, loganic acid, secologanin, and cinnamic acid
Introduction. Cancer is caused by changes in the human genome that increase cellular proliferation and disrupt normal routes and systems. Cancer is the second-leading cause of mortality globally [1]. One type of cancer, leukemia, is a category of hematological illnesses that cause the unregulated and abnormal proliferation of leukocytes [2].
Leukemia and the nervous system
Cancer is the leading cause of death in males under the age of 40 and
women below the age of 20 [3]. In 2020, approximately 60,530 new cases of leukemia were diagnosed, and 23,100 people were expected to die from this malignancy [4]. Chronic myeloid leukemia (CML), a kind of leukemia, is one of the blood illnesses known as "myeloproliferative syndromes." It is characterized by an increase in white blood cell production in the bone marrow. Some of these blood cells are abnormal; they are immature, meaning they are not fully developed when they reach the bloodstream. With an estimated frequency of one to two cases per 100,000 people, it accounts for around 15% of newly diagnosed leukemia in adults [5]. It was projected that over 9000 new CML cases would be detected in the United States in 2017, with approximately 1000 people dying from the disease [6]. The incidence of CML in the United States has risen from an estimated 25-30,000 cases in 2000 to a projected 80-100,000 cases in 2017 and will reach a peak of roughly 180,000 cases by 2030 [7]. CML is caused by a reciprocal translocation of chromosomes 9 and 22, resulting in the Breakpoint Cluster Region (BCR)-Abelson (ABL) oncogene, which produces a BCR-ABL fusion protein when translated [8, 9, 10]. The BCR-ABL chimeric oncogenic protein activates tyrosine kinase, causing leukemic cells to phosphorylate downstream effector molecules like Grb2, RAK, ROS, PI3K, JNK, STAT5, AKT, and Myc. This leads to uncontrolled cell proliferation via various signaling pathways [11, 12, 13]. BCR-ABL, a 210-kD protein with tyrosine kinase activity, is a possible target for discovering new anti-CML medicines [10]. Imatinib, a small-molecule inhibitor, has been effective in treating CML by targeting Abl kinase. Imatinib is a first-generation type II kinase inhibitor that targets the ATP-binding region of Abl kinase to reduce its activity [14]. Imatinib is both efficacious and well tolerated in patients with Ph+ CML. Imatinib outperformed interferon-α with cytarabine in stopping disease progression and obtaining haematological and cytogenetic responses in patients with newly diagnosed chronic-phase CML. It is still an effective therapy for all phases of the illness, particularly for newly diagnosed Ph+ chronic-phase CML, and is recommended as a first-line choice in European and US treatment recommendations [15]. But mutations in the Abl kinase domain can hinder Imatinib therapy by preventing the medication from binding to the binding site over time [16, 17]. Also, chemotherapy side effects and cancer cell resistance pose challenges for cancer treatment [18]. Therefore, we used Imatinib as a standard and designed the study for investigating potential anti-cancer drugs from the medicinal plant.
Plants are an infinite supply of several types of biologically active chemicals [19]. Phytochemicals like polyphenols, flavones, phenols, and flavonoids have been shown to have anti-cancer characteristics and can be utilized to treat many cancer types [20]. Studies (human, animal, in vivo, and in vitro) have indicated that phenolic compounds have favorable effects on plasma lipoproteins, oxidative damage, inflammatory indicators, platelet and cellular function, antibacterial activity, and bone health [21]. Olive tree (Olea europaea) leaves have been used in folk medicine to treat inflammation, allergies, diarrhea, and diseases like Alzheimer's, chronic fatigue syndrome, and osteoarthritis in the European Mediterranean, India, the Arabian Peninsula, and other tropical and subtropical regions [22]. Olive leaf extract (OLE) has unique phytocompounds that improve health results compared to olive fruit [23]. It has been proposed that the OLE plays an important protective role in inhibiting the proliferation of several cancer cell lines, including pancreatic, leukemia, breast, prostate, and colorectal [24]. Olive leaf contains bioactive chemicals that have been examined for their ability to suppress cancer progression and development, making it a promising target for future anticancer agents [1].
Drug development is a time-consuming and costly process with limited success and high attrition rates [25]. In silico approaches, such as structural bioinformatics, virtual screening, molecular dynamics, molecular docking, and comparative analysis of compounds with confirmed inhibitory effects, are highly cost-effective and versatile in drug discovery and biological research [26, 27, 28]. Molecular docking is a time-saving drug development approach that predicts ligand-receptor binding affinity and optimal orientation/pose during stable complex formation. Molecular docking can identify active chemicals that do not fit well into binding sites [29]. In this study, we utilized in-silico approaches such as (i) pharmacoinformatics to predict the toxicity, anticancer, and pharmacokinetic properties of the selected phytocompounds and (ii) molecular docking of 11 OLE phenolic compounds selected from Qais et al. [1] to identify promising candidates capable of interacting with and potentially inhibiting the activity of the BCR-ABL oncoprotein.
The aim of the study. This study aims to identify the potential lead phytocompounds from OLE to combat chronic myeloid leukemia in the coming future.
Materials and methods
Computer Configuration
The PC utilized in the current study was an Acer Aspire 7 laptop functioning Windows 11, with an AMD Ryzen 5 5500U CPU running at 2.1 GHz and 8 gigabytes of random access memory.
PASS Prediction
The anti-neoplastic, anti-carcinogenic, and anti-leukemic capabilities of the obtained phytocompounds were predicted using the PASS online service (https://www.way2drug.com/passonline/). The PASS (Prediction of Activity Spectra for Substances) algorithm assigns p values to similarity measurements based on whether anticancer activity is more or less likely [30]. With a false-positive rate of 0.05, it may achieve up to 65% accuracy. We used the PubChem website to obtain the canonical SMILES of chemicals for our investigation.
ADMET Analysis
Analysis of ADMET (absorption, distribution, metabolism, excretion, and toxicology) characteristics plays a crucial role in rational drug design and development. These parameters provide valuable information about the toxicity and effectiveness of drugs. An effective drug must have appropriate solubility, permeability to the tissue, and stability [31]. In silico methods are less expensive and faster than experimental methods for estimating ADMET values. We used the SwissADME online server (http://www.swissadme.ch/) to calculate the needed criteria for the desired compounds in ADME [32]. To examine the toxicological features of chosen chemicals, we used the free online web application AdmetSAR (http://lmmd.ecust.edu.cn/admetsar2/) [33].
Protein preparation
The structure of the BCR-ABL (PDB ID: 5MO4) protein has been obtained from the PDB (protein data bank) online website (https://www.rcsb.org/) at a resolution of 2.17Å. The protein's structure was available in PDB format. The protein structure is translated from PDB to PDBQT format using Autodock tools (1.5.6). Before conversion, the water molecules from the protein structure were removed. We used polar hydrogen atoms and repaired missing atoms. Kollman charges were incorporated into the protein structure, and they were dispersed evenly. The resulting PDBQT structure was saved [34, 35].
Ligand preparation
Ligand structures consisting of 11 phenolic OLE compounds and the standard anti-CML drug Imatinib have been obtained from the PubChem online website (https://pubchem.ncbi.nlm.nih.gov/). The 3D structures of the ligand include Verbascoside (PubChem CID: 5281800); Uvaol (PubChem CID: 92802); Loganic acid (PubChem CID: 89640); Secologanin (PubChem CID: 161276); Ferulic acid (PubChem CID: 445858); Hydroxytyrosol (PubChem CID: 82755); Gallic acid (PubChem CID: 370); Coumaric acid (PubChem CID: 637542); Homovanillyl alcohol (PubChem CID: 16928); Cinnamic acid (PubChem CID: 444539); Tyrosol (PubChem CID: 10393); and Imatinib (PubChem CID: 5291). The structures of the ligands were in SDF format. Using OpenBabel software, the SDF format can be converted into the PDB format [36]. The PDB format of the ligand structures is converted to the PDBQT format by using Autodock tools (1 5.6) [4].
Molecular docking
In order to identify new potent inhibitors of tyrosine kinase, docking simulation was done, and the calculation of binding energies was performed using Autodock Vina. Autodock Vina is open-source software for molecular docking in silico. Dr. Oleg Trott from The Scripps Research Institute's molecular graphics department conceived and implemented the system [37]. This was done to forecast the binding energy scores for the interactions between the targeted protein ligands. The Autodock Vina program now uses the identified binding sites of amino acid residues from specific proteins. The docked confirmations with the highest fitness score were used to analyze the binding mode. Firstly, the grid box with the following dimensions was set to cover the protein structure with the following dimensions in Å: center (X = -33.722, Y = 19.579, Z = -8.128), dimension (X = 74, Y = 68, Z = 70) with an exhaustiveness of 8. After running the Autodock Vina software, binding energies were measured, recorded, and displayed. Finally, Discovery Studio Visualizer v21 was used to analyze docking and generate images of the protein-ligand complexes [38, 39].
Results
PASS Prediction
The PASS Online program was used to identify probable cellular targets of OLE phytocompounds before docking analysis.
We used PASS Online to predict anticancer efficacy based on three primary parameters: anti-carcinogenic, anti-neoplastic, and anti-leukemic. The PASS Online server predicts complex biological activities and provides indicators of activity or inactivity [30]. There is a probability of either biological activity (Pa) or biological inactivity (Pi). Table 1 shows the projected anticancer characteristics of OLE compounds.
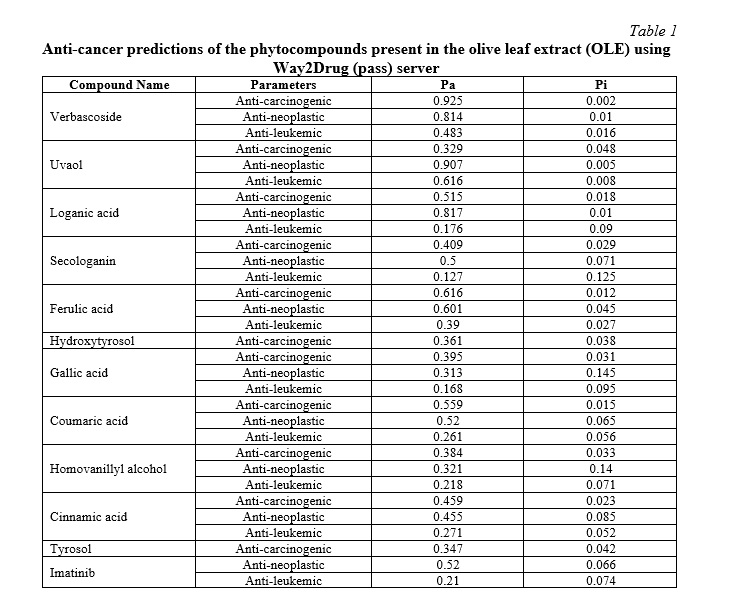
ADMET Analysis
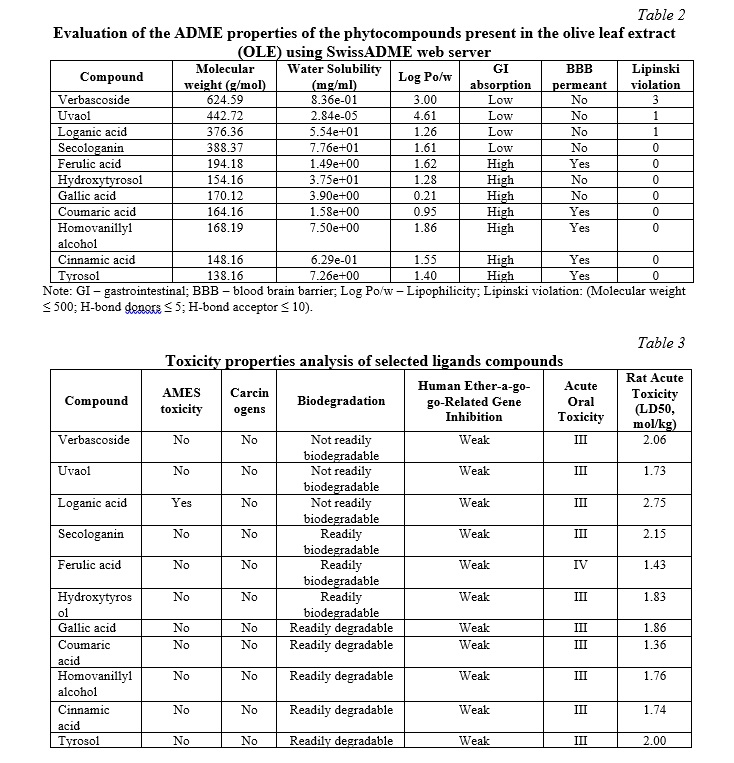
SwissADME estimates ADME and the drugability potency of substances using biological characteristics. The ADME characteristics of the chosen compounds indicated drug-like qualities and potential therapeutic applications. The ADME results are reported in Table 2. Based on their toxicity characteristics, the investigated chemicals were determined to be non-AMES harmful and non-carcinogenic. Table 3 summarizes the toxicological features of the identified phytochemicals.
Molecular Docking
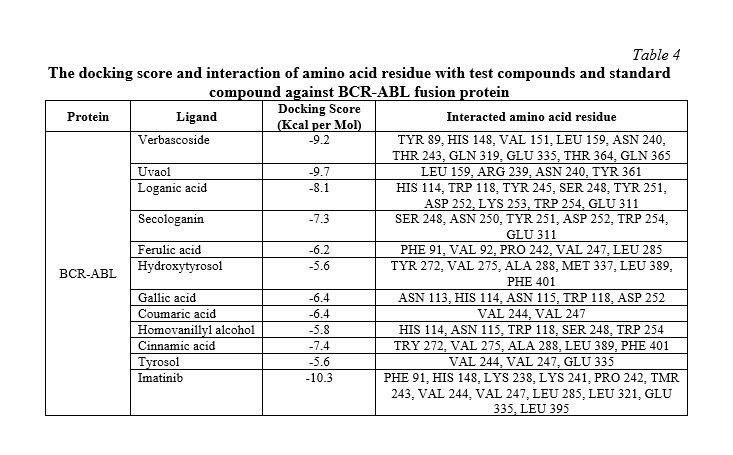
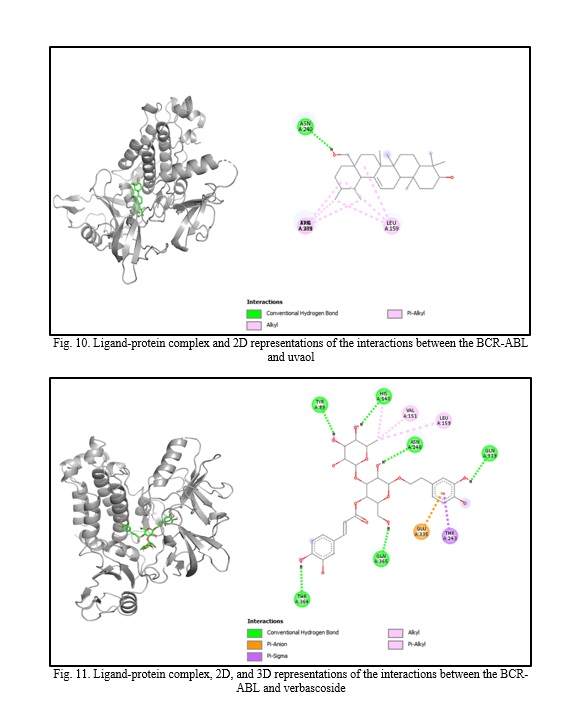
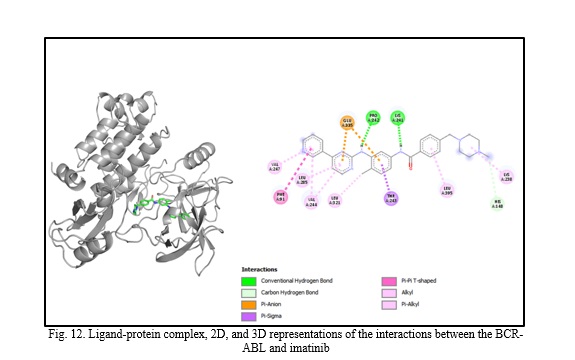
In silico molecular docking experiments demonstrate that verbascoside, uvaol, loganic acid, secologanin, and cinnamic acid have a high binding affinity to the BCR-ABL protein, with binding energies of -9.2, -9.7, -8.7, -7.3, and -7.4, respectively, which are extremely near the binding affinity of the standard medication Imatinib, which has a binding energy of -10.3 Kcal/mol. The interactions between the chemicals and the amino acid residues are pretty interesting. Table 4 lists the amino acid residues to which the compounds bind, as well as the ligands' binding energies. Figure 1 to Figure 12 demonstrate ligand-protein and 2D interactions between the BCR-ABL and desired compounds.
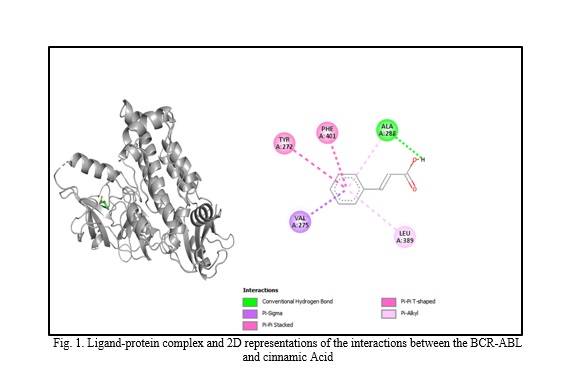
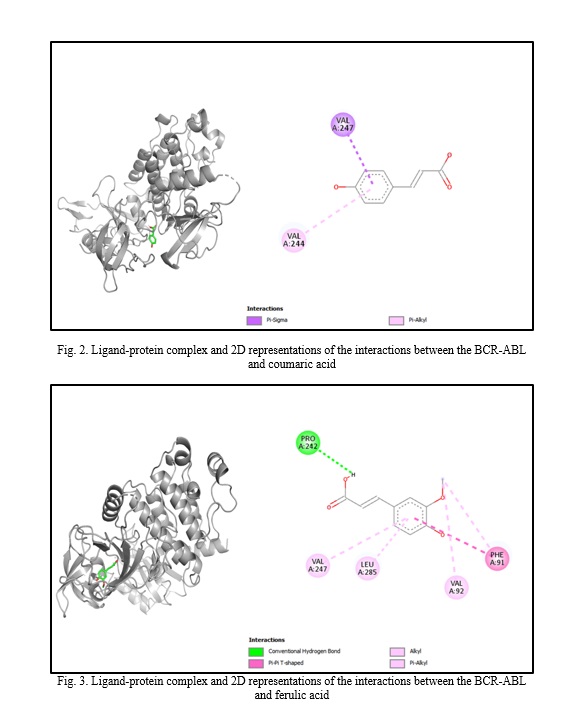
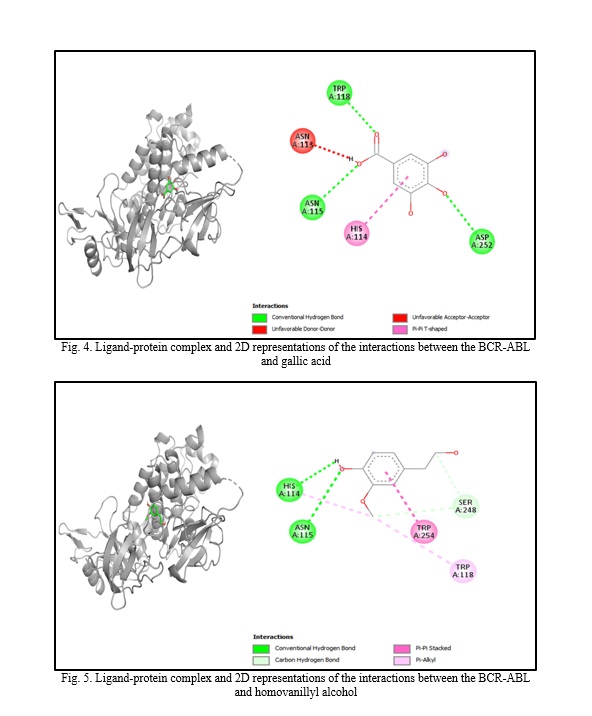
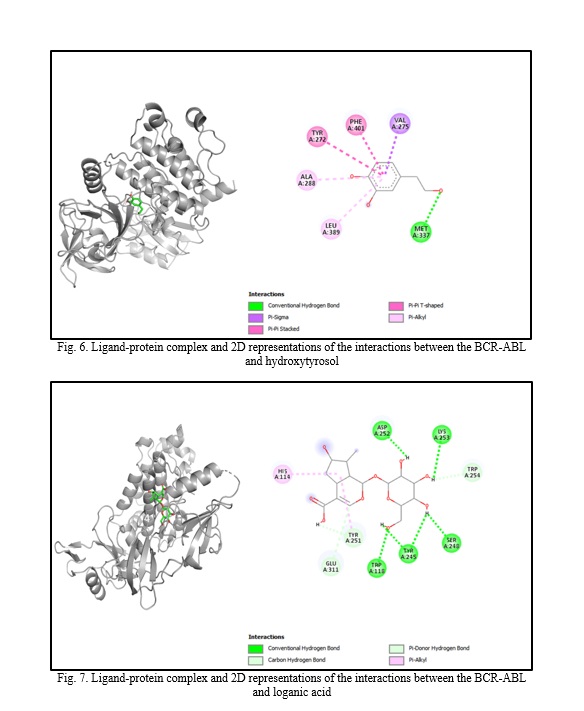
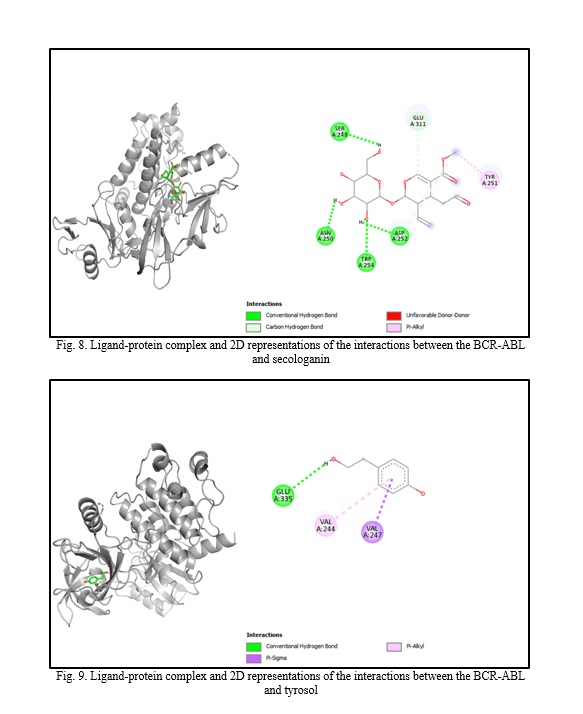
Discussion. Medicinal plants are increasingly used to cure deadly illnesses due to their high concentration of bioactive phytochemicals and safety compared to synthetic drugs. These active chemicals have been shown to cure many infections and illnesses, including cancer. Scientists are exploring natural sources for active compounds to treat cancer due to the significant negative consequences of current therapy choices [33]. Plants are a reliable source of novel anticancer medications, accounting for almost 60% of all currently available treatments [40]. Large pharmaceutical corporations invest heavily in developing medications that target cancer cells, as the demand for safer and more effective chemotherapy medications stems from serious side effects and resistance to current treatments [41, 42]. Identifying natural pharmacological compounds with target-specific action is an effective technique for developing novel drugs due to their minimal toxicity [14]. When developing an active component, it's vital to consider bioactivity, pharmacokinetics, and toxicology. Many medication candidates are unable to advance to the final stage of drug design owing to failing to fulfill certain requirements [43]. Computer-aided drug design (CADD) is a widely used, sophisticated technology for drug design that may reduce prices and save time. Scientists use pharmacokinetic analysis and molecular docking to identify the most successful medication candidates [44]. In silico analysis is preferred over animal models for validating drug candidates due to their high cost, difficulty, and time commitment. In silico investigation of pharmacokinetic characteristics and toxicological parameters may be completed quickly and accurately [45]. Thus, in this work, we employed in silico molecular docking, ADME analysis, and PASS prediction to identify drug-like natural compounds for the treatment of chronic myeloid leukemia.
We used PASS online to predict anticancer activity, taking into account three primary parameters: anti-carcinogenic, anti-neoplastic, and anti-leukemic. All substances had Pa values greater than Pi, indicating a higher chance of biological activity than inactivity. Interestingly, all the compounds have significantly higher Pa values compared to Pi values. This indicates that these chemicals are more likely to inhibit the BCR-ABL protein.
The SwissADME website predicts ADME qualities for phytocompounds, indicating relevant pharmacological properties (see Table 2). To increase the quality of a medicine, it must have good toxicological properties, such as no AMES toxicity, non-carcinogenicity, and mild hERG inhibitors [46, 47, 48]. The experimental compounds had comparable findings, as shown in Table 3. AMES toxicity evaluates the mutagenic potential of substances [49]. Loganic acid was the only investigated chemical that exhibited AMES toxicity. The toxicological examination of the chosen compounds suggests that the majority of phytocompounds are not harmful.
A molecular docking study was conducted to identify the optimal interaction between the BCR-ABL protein and phytocompounds. This study utilized the Autodock Vina software to dock the chosen OLE phenolic phytocompounds.
Docking using Autodock Vina yielded nine optimal conformations, with the lowest binding energy being selected. Table 3 shows the binding energies and interacting amino acid residues. Docking scores indicate binding affinities. A pharmaceutical candidate with a lower binding energy is always favored [50]. The free energy created during the formation of the receptor-ligand complex indicates the ligand's affinity for the receptor. A strong affinity between a ligand and a receptor leads to a lower free energy value and docking score. If the affinity is small, the free energy value or docking score rises [51]. The compounds have high binding affinity for BCR-ABL, with binding energies ranging from -5.6 to -10.3 kcal/mol.
Verbascoside, uvaol, loganic acid, secologanin, and cinnamic acid were the top five compounds discovered based on binding energy, binding site, pharmacokinetics, ADME characteristics, and projected anticancer activity. The first three exhibited an extremely close binding affinity to Imatinib. Later two demonstrated medium proximity to the binding affinity of Imatinib. Several studies have also confirmed the anti-leukemic and anti-cancer properties of verbascoside [52], uvaol [53], loganic acid [54], and cinnamic acid [55], which were discovered through our current in silico investigations. While these five compounds violated several in silico drug development requirements, there are other examples of similar infractions among current medications [56]. After reviewing all of the data, it is clear that certain phenolic compounds from OLE are effective anti-leukemic phytocompounds that can be employed in future research to develop new medications.
Future implication and limitations of study
Our findings have interesting implications for future in vivo and clinical studies, potentially leading to the development of new treatment drugs and combination therapies for chronic myeloid leukemia (CML). However, the study's in-silico nature restricts its immediate usefulness, as the effectiveness and safety of the phenolic compounds must be verified in experimental and clinical trials. Further studies might include in vitro and in vivo studies to establish the physiological consequences of these findings.
Conclusion. The study aims to identify the therapeutic potential of olive plant-based phenolic compounds against chronic myeloid leukemia. In silico data indicate that verbascoside, uvaol, loganic acid, secologanin, and cinnamic acid have significant anti-cancerous action and therapeutic promise against CML.
The findings suggest that phytochemicals might be effective in the discovery of potent anti-leukemic drugs.
Financial support
No financial support has been provided for this work.














Reference lists