
Clinical characterization of a child with de novo partial trisomy 9p24-9q12
Abstract
Background: Partial trisomy 9p is considered the fourth most frequently reported trisomy in live births, following trisomies 21, 18, and 13. This relatively high incidence is observed, probably, due to the limited number of genes involved within the 9p24-9q12 region. With a frequency of less than 1 in 1,000,000 live births, it is classified as a rare disease. The aim of the study:To describe the phenotypic characteristics of a child with de novo partial trisomy 9p and compare them with previously documented cases in the international literature. Materials and methods: A descriptive case report of a 4-year-old male patient with global neurodevelopmental delay and dysmorphic features evaluated at the William Soler University Pediatric Hospital. Clinical, anthropometric, dermatoglyphic, imaging (computed tomography, echocardiography, ultrasound), and audiometric (brainstem auditory evoked potentials) assessments were carried out. Cytogenetic analysis was performed on peripheral blood lymphocyte cultures using the synchronization method, with high-resolution GTG-banded karyotyping. Fluorescence in situ hybridization (FISH) was used to characterize the supernumerary chromosome with VYSIS (Abbott) probes. The clinical and cytogenetic findings were compared with previously published cases. Results: A 4-year-old male patient presented with global neurodevelopmental delay and distinctive facial dysmorphisms. At birth, he exhibited cyanosis and an absence of the crying and sucking reflexes. He was subsequently noted to have severe neurodevelopmental delay from the first month of life, and initially presented with hypotonia, which improved following physical therapy. His social development was poor, and language development was nearly non-existent. Physical examination revealed low height and weight for his age, alongside multiple facial dysmorphic features. He was also diagnosed with a Dandy-Walker malformation, pulmonary valve dysplasia, and an 8 mm atrial septal defect. Karyotyping identified a large supernumerary chromosome, which, via GTG banding, was characterized as partial trisomy 9p, consistent with the chromosomal formula: 47,XY,+del(9)(q12q34.3). Cytogenetic analysis of both parents showed a normal constitution. Conclusion: The patient's clinical findings were consistent with those reported in the literature, which, in conjunction with conventional cytogenetic studies, led to the diagnosis of de novo partial trisomy 9p
Keywords: Karyotype, Trisomy, Neurodevelopmental Disorders, Rare Diseases, Chromosomes, Human, Pair 9
Introduction. Partial trisomy 9p is internationally recognized as a rare disease. As of 2023, 225 cases have been described in the scientific literature, potentially involving regions of varying length across the short arm of chromosome 9. Owing to the relatively low proportion of genes involved in this region, 9p trisomy is the fourth most frequently described autosomal trisomy in live-born infants, following trisomies 21, 18, and 13. Its prevalence is estimated to be less than 1 in 1,000,000 live births [1-4].
The typical dysmorphic features observed are influenced by the specific portion of chromosome 9 involved. Trisomy restricted to the 9pter-p11 region often dictates the classic craniofacial phenotype, whereas trisomy of 9pter that extends into the long arm (q11−13) is generally associated with severe skeletal and cardiac defects [1]. Several studies have further characterized the critical region for the classic phenotype as 9p22/p23. Consequently, some researchers consider the 9p22 area a critical region responsible for many of the cardinal symptoms of 9p trisomy [3]. However, this correlation is not universally observed, as cases described in the literature with duplications of this specific region have exhibited only moderate clinical features and symptoms [1].
The aim of the study. To characterize the phenotypic features of a child with a de novo complete 9p trisomy and to compare these findings with existing international literature reports.
Materials and Methods
I. Patient Data
The patient is a 4-year-old male presenting with global neurodevelopmental delay and facial dysmorphisms. He is the product of a non-consanguineous couple. The mother reported three recurrent spontaneous abortions. No other relevant family history was noted.
II. Perinatal History
Pregnancy:
- Complicated by recurrent urinary tract infections.
- Alpha-fetoprotein screening and all prenatal ultrasounds were reported as normal.
Delivery:
- Dystocic delivery at 41 weeks of gestation.
Complications:
- Premature rupture of membranes, apparent oligohydramnios, and fetal distress.
Birth:
- Presented with a fractured clavicle.
Birth Anthropometry:
- Weight: 3104 g
- Head Circumference: 34 cm
- Length: 50 cm
- Apgar Scores: 1/7.
Neonatal Period:
- Did not cry or suckle at birth, exhibiting cyanosis.
- Required admission to the Intensive Care Unit for 3 days, followed by intermediate care for pneumonia.
III. Current Development and Evaluation (at 4 years)
Neurodevelopment:
- The patient has shown severe neurodevelopmental delay since the first month of life.
- Initial hypotonia improved with physical therapy.
- Currently exhibits poor social development and severely limited (almost non-existent) expressive language.
Current Anthropometry (at 4 years old):
- Weight: 14 kg (10th-25th Percentile)
- Height: 96.5 cm (10th-25th Percentile)
- HC: 49 cm (25th Percentile)

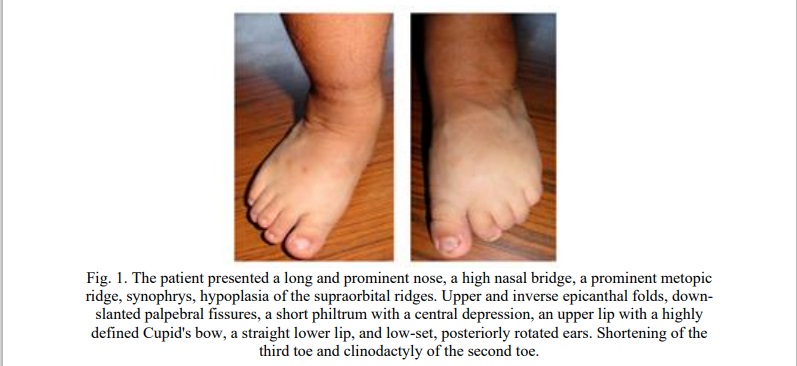
IV. Bone Age and Skeletal Malformations
Bone Age:
- No ossification was observed in the carpal bones, corresponding to an estimated bone age of 0 to 3 months.
Phalangeal Abnormalities:
- The phalanx of the fifth digit was undefined on both hands.
- The second phalanx of the first digits (thumbs) was small for the expected age.
V. Imaging Studies and Major Diagnoses
Computed Tomography (CT) Scan:
- Diagnosis of a Dandy-Walker malformation was established.
Echocardiogram:
- Revealed pulmonary valvular dysplasia and an atrial septal defect (ASD) measuring 8 mm.
Abdominal Ultrasound (US) and Ophthalmology:
- Abdominal US: Normal.
- Ophthalmology: Normal.
VI. Specific Findings
Brainstem Auditory Evoked Potentials (BAEP):
- Mild hearing loss was detected.
Soft Tissue US (Sacral Region):
- At the level of the sacral dimple, a hyperechogenic tract was observed extending from the skin and appearing to reach the sacrum. This tract showed a tendency to deviate toward the left.
VII. Dermatoglyphics (Palmar Impressions) (See Figure 2)
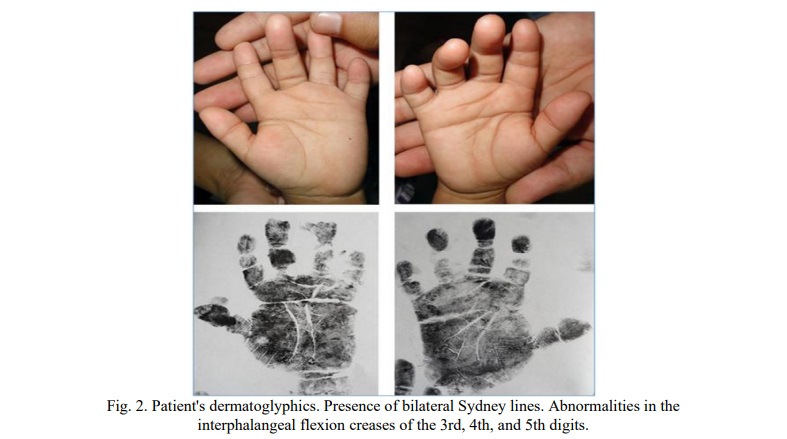
The dermatoglyphic findings are suggestive of chromosomal abnormalities (chromosomopathies).
General Patterns:
- Predominance of ulnar loops.
Sydney Line:
- Presence of a bilateral Sydney Line (a finding with a reported value of <0.5 in the Cuban population).
Creases and Syndactyly:
- Abnormalities were noted in the interphalangeal flexion creases of digits 3, 4, and 5.
- Type I syndactyly pattern was present in the 3rd and 4th interdigital spaces, which is considered uncommon for this area.
Triradii:
- Presence of an interdigital triradius type 2 in the second space of the right hand.
- An axial triradius on the right hand was located at t′, extremely high, nearly at t′′.
Thenar Region:
- True figures (patterns) were present on the left hand and a vestigial pattern on the right hand.
Results. Chromosomal analysis was performed on peripheral blood lymphocyte cultures using the synchronization method. Blood samples were prepared for high-resolution karyotyping analysis employing trypsin followed by Giemsa staining to obtain a standard GTG banding pattern. The GTG-banded chromosomal analysis initially revealed a result of 47,XX,+mar.
Given the presence of a supernumerary chromosome, the sample was further analyzed using the Fluorescence In Situ Hybridization (FISH) technique with the probes available in the laboratory. The following VYSIS (ABBOT) probes were utilized: LSI SRNPN spectrum green/CEP 15(D15Z1) spectrum red/LSI PML spectrum orange, which is specific for the critical region of Prader-Willi and Angelman syndromes. The signal pattern for this probe was normal. VYSIS Centromeric Probes (CEP) for chromosomes X, 18, and Y. LSI probes for chromosomes 21, 13, 22, and 7. In all of the above FISH assays, no chromosomal signal alteration was observed. Specific probes for any region of chromosome 9 were not available.
Based on the GTG banding pattern, this fragment corresponds to an extra short arm of chromosome 9, leading to the final karyotype designation: 47,XY,+del(9)(q12q34.3) (Fig. 3).
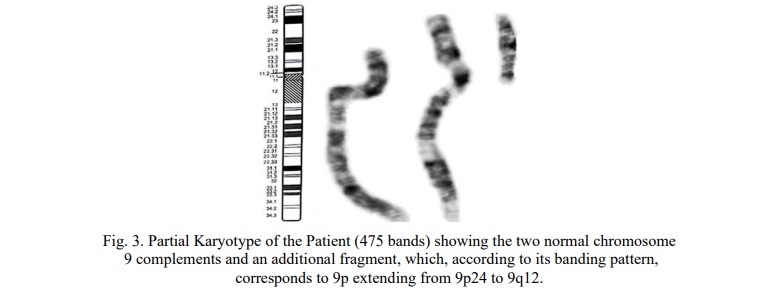
The maternal karyotype was determined to be 46,XX and the paternal karyotype was 46,XY, suggesting that the abnormality is likely a de novo event. Gonadal mosaicism in either parent could not be definitively ruled out.
Discussion. The majority of cases involving partial 9p trisomy arise from an imbalance during gametogenesis in one parent who carries a reciprocal translocation between chromosome 9 and another chromosome [5]. In such cases, the precise clinical delineation of the syndrome may be complicated by the partial monosomy of the other chromosome involved in the parental translocation. Indeed, the phenotypic heterogeneity observed in 9p trisomy may be influenced by this co-occurring event, in addition to the size of the segment involved in the partial trisomy [6, 7].
When 9p trisomy is reported as apparently de novo within a family, determining the underlying mechanism remains challenging. In the case analyzed herein, the extra chromosome possesses a breakpoint at 9q12 within the pericentromeric region of the long arm. This region is notably rich in low-copy repeat (LCR) segments, which predispose it to Non-Allelic Homologous Recombination (NAHR). This mechanism can generate multiple polymorphic variants of chromosome 9 [8, 9] and also renders the region prone to rearrangements, such as reciprocal translocations between chromosome 9 and other human chromosomes due to sequence homologies at the breakpoints [10].
Phenotype-Genotype Correlation
According to Wright et al. [10], the 9p24.3−p13.1 region, which is similar in size to the segment reported in our case, encompasses 182 genes. Of these, 48 genes are described in OMIM as having dominant or recessive inheritance patterns. This gene set includes VLDLR (OMIM #192977), JAK2 (OMIM #147796), NFIB (OMIM #600728), PLAA (OMIM #603873), TEK (OMIM #600221), VCP (OMIM #601023), RUSC2 (OMIM #611053), NPR2 (OMIM #108961), PAX5 (OMIM #167414), and CNTNAP3 [11].
Several of these genes may be implicated in the patient's phenotypic characteristics, including TEK, NFIB, and NPR2, which have been linked to conditions such as macrocephaly associated with intellectual disability, short stature, and neurodevelopmental disorders [12, 13, 14]. Additionally, the genes PAX5 and CNTNAP3 are described as playing roles in central nervous system development and the learning process [15].
Other authors have hypothesized that when the partial trisomy extends to involve the centromere and part of the long arm (e.g., 9pter→q11−13), the phenotype often includes skeletal anomalies and cardiovascular defects in addition to the features previously reported, consistent with the presentation of the patient in the current study [4, 5]. Some literature reports suggest that the candidate region for Dandy-Walker Syndrome in partial 9p trisomy is located when regions of the long arm, up to 9q22.2, are included [16, 17, 18].
The detailed clinical findings described in our patient align with many of the core features of 9p trisomy, such as short stature, delayed bone development, a large nose with a bulbous tip, downturned mouth corners, brachydactyly, clinodactyly, language delay, and bilateral simian crease, which are documented in 90-99% of patients with this syndrome [4]. Other characteristics, including low-set and deformed ears, phalangeal hypoplasia, hypotonia, low birth weight, and intellectual disability, are observed in 60-90% of patients [4]. This meticulous clinical description is highly concordant with the cytogenetic findings. To further corroborate this cytogenetic result, a patient microarray study would have been necessary to more precisely delineate the partial trisomy of chromosome 9.
Notably, among the less frequently observed phenotypic characteristics are the pulmonary valvular dysplasia and an 8 mm atrial septal defect, alongside the Dandy-Walker skeletal malformation. As previously emphasized, these anomalies are typically associated with 9p trisomy that includes sections of the long arm, which lends further credence to the cytogenetic finding.
Limitations of the Study
The diagnosis was obtained using conventional cytogenetics. Molecular methods for a more accurate delineation of the extra chromosomal fragment were not available. Therefore, potential inaccuracies regarding the precise location of the breakpoint on the long arm of chromosome 9 may exist.
Conclusion. The clinical findings in the patient were consistent with those described in the literature. These findings, when complemented by conventional cytogenetic studies, permitted the diagnosis of de novo complete 9p trisomy.
Financial support
No financial support has been provided for this work.
Conflict of interests
The authors have no conflict of interest to declare.














Reference lists